Mitochondrial energy production is essential for life, and mitochondrial dysfunction can impact many aspects of health.
This article explains one of the enzymes at the heart of the mitochondrial energy production cycle, pyruvate dehydrogenase. Members will see their genotype report below. Consider joining today.
What is pyruvate dehydrogenase?
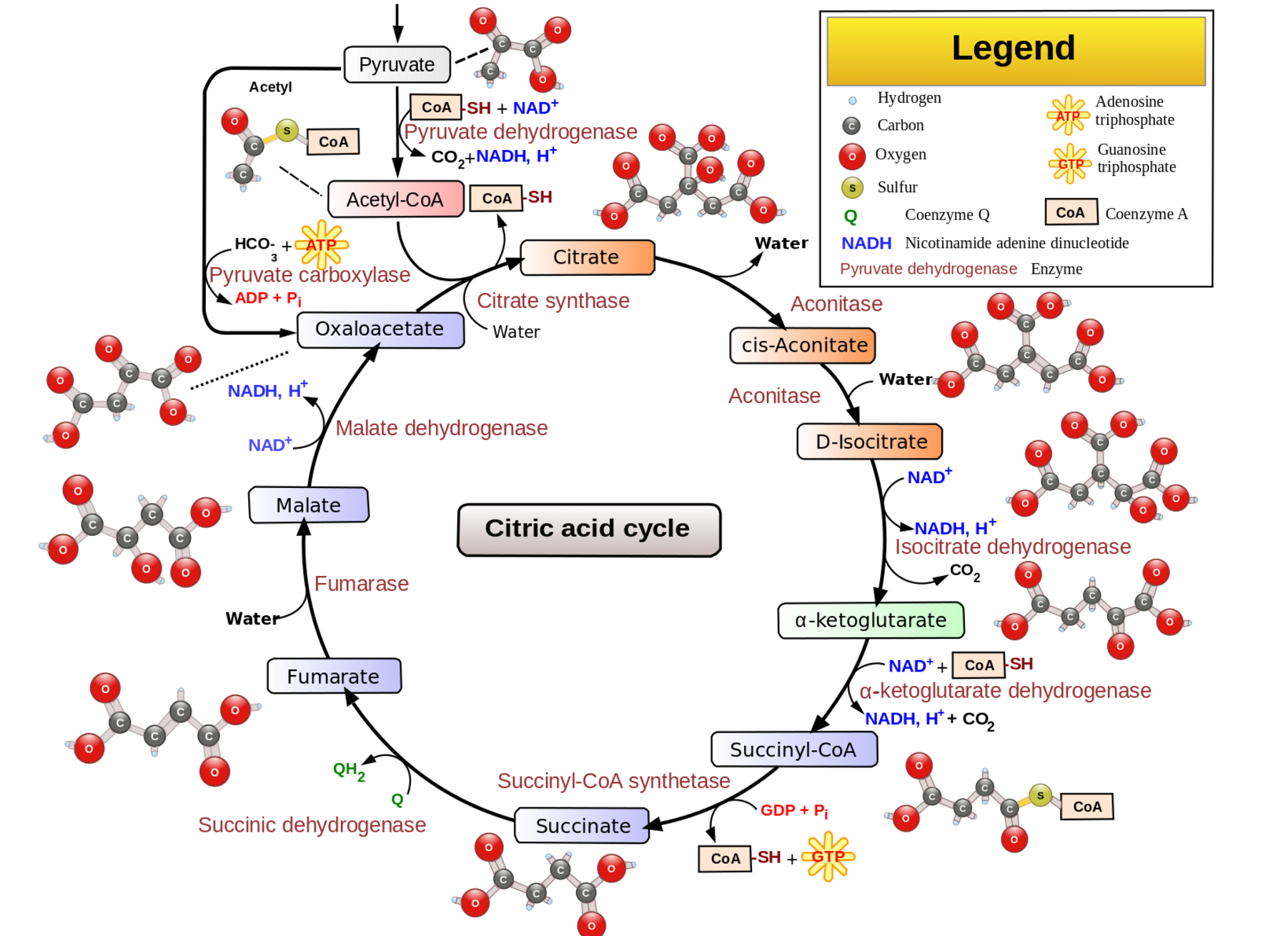
Pyruvate dehydrogenase is an enzyme vital to the production of cellular energy in the mitochondria. It acts as a catalyst in the conversion of pyruvate into acetyl-CoA, which is used in the citric acid cycle (Kreb’s cycle) in cellular respiration and the production of ATP.
Thus, pyruvate dehydrogenase is essential, and deficiencies of pyruvate dehydrogenase can be devastating.
Quick background on mitochondrial energy production:
Mitochondria are the organelles responsible for creating most of the ATP used by cells. I say “most”, because glycolysis in the cytosol can also produce a little ATP.
ATP stands for adenosine triphosphate, which is the molecule used for energy in cellular processes in all life forms. In a nutshell, the phosphate bond is easily broken and gives up energy when adenosine triphosphate (ATP) becomes adenosine diphosphate (ADP).
In the mitochondria, the citric acid cycle converts pyruvate into ATP. Additionally, more ATP is created in the inner membrane of the mitochondrium through oxidative phosphorylations.
What is pyruvate?
Pyruvate is a small molecule created from glucose in glycolysis (breaking apart of glucose for energy). Pyruvate is then used in the citric acid cycle to create energy, or it can be converted into glucose again, via gluconeogenesis. Additionally, pyruvate can be converted into lactic acid during fermentation.
Pyruvate Dehydrogenase Deficiency:
Babies born with two copies of mutations in the pyruvate dehydrogenase genes can have pyruvate dehydrogenase deficiency. This causes a buildup of lactic acid, which causes neurological and developmental problems. This rare genetic disease is often lethal in childhood. [ref]
But what happens if you just have one copy of a mutation linked to pyruvate dehydrogenase deficiency? Or what if you have other conditions that impact the use of pyruvate for mitochondrial energy production?
Link to CFS/ME:
A December 2016 report looking at the metabolites in Chronic Fatigue Syndrome patients found indicators showing impairment of pyruvate dehydrogenase. It concludes: “These findings are in agreement with the clinical disease presentation of ME/CFS, with inadequate ATP generation by oxidative phosphorylation and excessive lactate generation upon exertion.”
Rare pyruvate dehydrogenase deficiency mutations: Genotype report
PDHA1 Gene (mainly 23andMe v4)
Members: Log in to see your data below.
Not a member? Join here.
Why is this section is now only for members? Here’s why…
Member Content:
Why join Genetic Lifehacks?
~ Membership supports Genetic Lifehack's goal of explaining the latest health and genetics research.
~ It gives you access to the full article, including the Genotype and Lifehacks sections.
~ You'll see your genetic data in the articles and reports.
Join Here
Lifehacks:
Diet and Nutrients:
Thiamine and magnesium are cofactors of pyruvate dehydrogenase. Thus, getting enough thiamine (vitamin B1) as well as magnesium is important. [ref]
In a recent case study, supplemental thiamine along with dietary restrictions reversed muscle weakness in a boy with pyruvate dehydrogenase deficiency. [ref]
Ketogenit diet:
An animal study found that a ketogenic diet may be helpful in the prenatal development of a pyruvate dehydrogenase deficient mouse.[ref]
Cannabinoids:
An interesting study recently looked at the mitochondrial cannabinoid receptors and their role in skeletal muscle metabolism including the gene expression of PDHA1. The study concludes: “The activation of mtCB1 receptors may participate in the mitochondrial regulation of the oxidative activity probably through the relevant enzymes implicated in the pyruvate metabolism, a main substrate for TCA activity.”[ref]