With COVID-19 all over the news, many people have now heard the term ARDS bandied about. Acute respiratory distress syndrome (ARDS) is a very serious condition that a minority of people with COVID-19 develop. This is the reason, though, that everyone is discussing ‘flattening the curve’ as a way to decrease the number of patients on ventilators in the ICU at one time.
This article explains what happens to the body in ARDS, and it goes into the genetic variants that increase or decrease the risk of ARDS (due to all causes – not just COVID-19). ARDS is a ‘syndrome’ and thus a collection of symptoms rather than a disease.
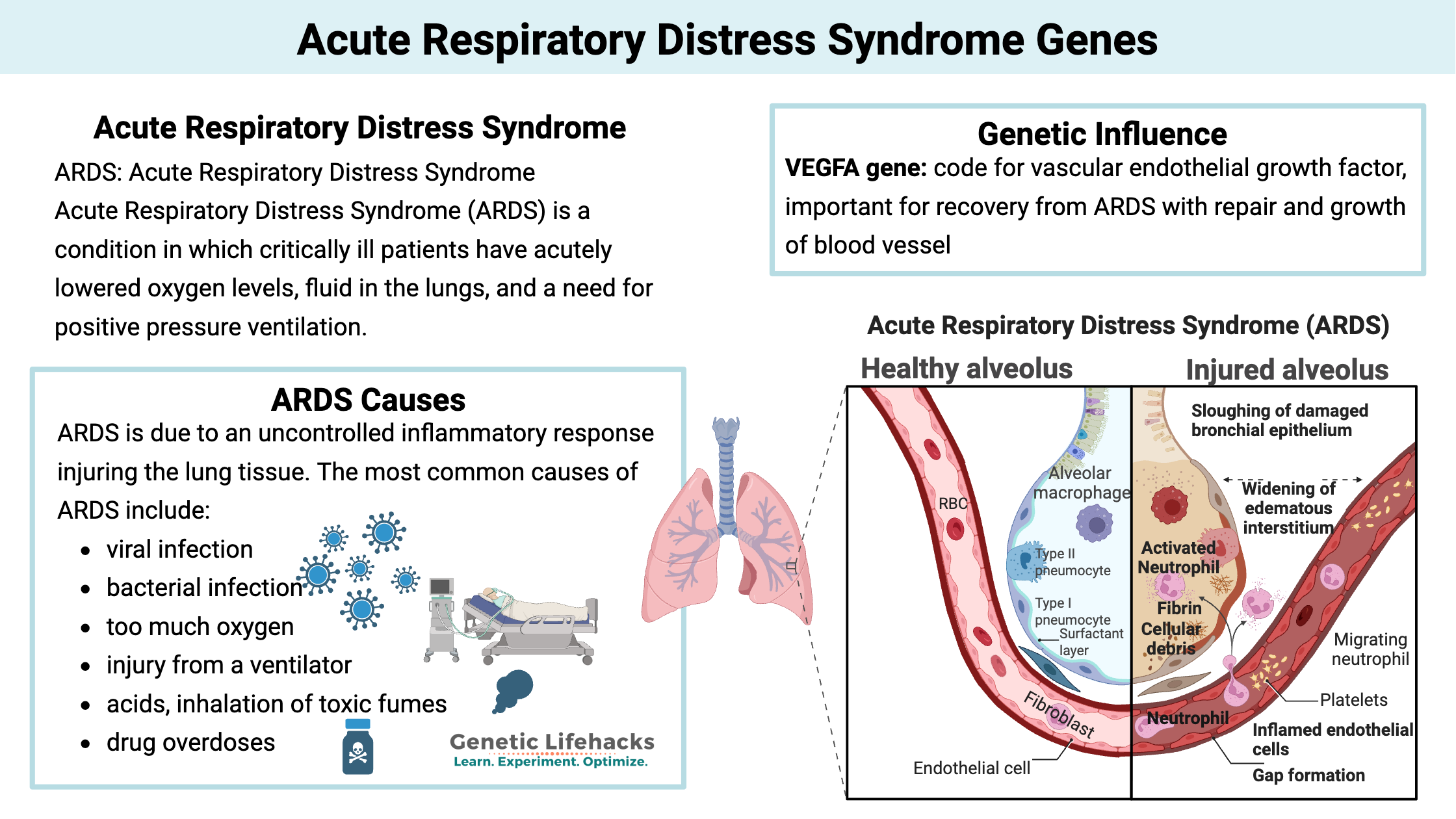
ARDS: Acute Respiratory Distress Syndrome
Acute Respiratory Distress Syndrome (ARDS) is a condition in which critically ill patients have acutely lowered oxygen levels, fluid in the lungs, and a need for positive pressure ventilation. ARDS is due to an uncontrolled inflammatory response injuring the lung tissue.[ref]
This syndrome was first defined in 1967, with updates periodically to the diagnosis criteria. The 2012 ARDS diagnosis definition includes that respiratory failure occurs without being explained by heart function and that oxygenation is needed at specific levels.[ref]
What are the causes of ARDS?
The most common causes of ARDS are pneumonia, sepsis, aspiration, and major trauma. The mortality rate is high for people with ARDS – from 35 – 36% mortality, depending on the severity.
Inside the lungs, the major airways (bronchi) continue to branch off into smaller and smaller bronchi before becoming bronchioles. At the end of all the branching, these bronchiole air passages progress into alveoli – little air-filled sacs surrounded by capillaries. This is the site of gas exchange for oxygen and carbon dioxide. Oxygen enters the bloodstream, and carbon dioxide leaves.
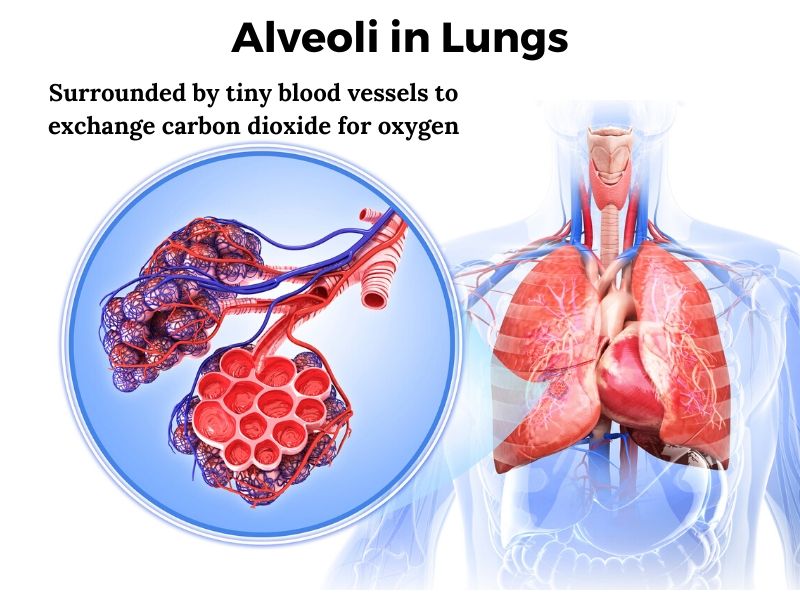
Two types of epithelial cells line the alveoli, forming a very tight barrier that only allows the passage of carbon dioxide and oxygen gases. These important tight junction bonds between the cells are created from specific proteins.
In ARDS, the epithelial cells lining the alveoli no longer remain tightly bound together, allowing them to fill with fluid that leaks in from surrounding tissue. Once the alveoli fill up with the leaked fluid, the gas exchange can’t happen. This causes both a decrease in oxygen and a buildup of carbon dioxide.[ref]
This is why just giving a patient more oxygen when they have ARDS doesn’t do any good. The alveoli are no longer able to make the gas exchange with the capillaries. Mechanical ventilation is needed in order to reopen and stabilize the alveoli.
So what causes the epithelial cells lining the alveoli to be damaged and no longer tightly joined together?
There are a variety of different causes including:[ref]
- viral infection(such as SARS-CoV2 as well as other pneumonia-causing viruses)
- bacterial infection (such as pneumonia-causing bacteria)
- too much oxygen (hyperoxia)[ref]
- injury from a ventilator
- acids, inhalation of toxic fumes
- drug overdoses
- trauma
When the epithelial cells are initially damaged, the body produces an immune response – sending in neutrophils and monocytes in to kill the pathogens. But this inflammatory response also damages the epithelial cells further. This is the second wave of damage that occurs in the lungs, due to our own immune response.
Capillaries leaking fluid: During ARDS, the capillaries surrounding the alveoli also play a role. The immune response causes increased permeability in the blood vessels, allowing plasma to leak out. This causes increased fluid around the alveoli, which combines with the decrease in the epithelial cell barrier to flood the alveoli with fluid.
Finally, there is a long road to healing. The inflammatory response must be tamped down, and the damaged lung tissue must be repaired. The recovery period can be extensive, with the patients continuing to have shortness of breath with exertion.[ref]
What makes someone more susceptible to ARDS?
Factors that increase the susceptibility to ARDS include:[ref][ref]
- alcohol abuse
- smoking
- air pollution
- age
- immunosuppression
- right ventricular dysfunction
What can genetics tell us about ARDS?
Looking at the genetic variants that increase the risk for ARDS is one way of knowing the pathways involved in this syndrome. Researchers can measure what is going on in the blood when someone has ARDS, and they can look at animal research models to see what goes on specifically in the alveoli. But genetics research can also show exactly which inflammatory molecules and which cellular junction molecules are involved both in the initial cascade of symptoms as well as the final steps needed to turn the corner and heal. Certain genetic variants are linked to an increased risk of having ARDS, and other variants are linked to a poorer outcome if someone has ARDS.
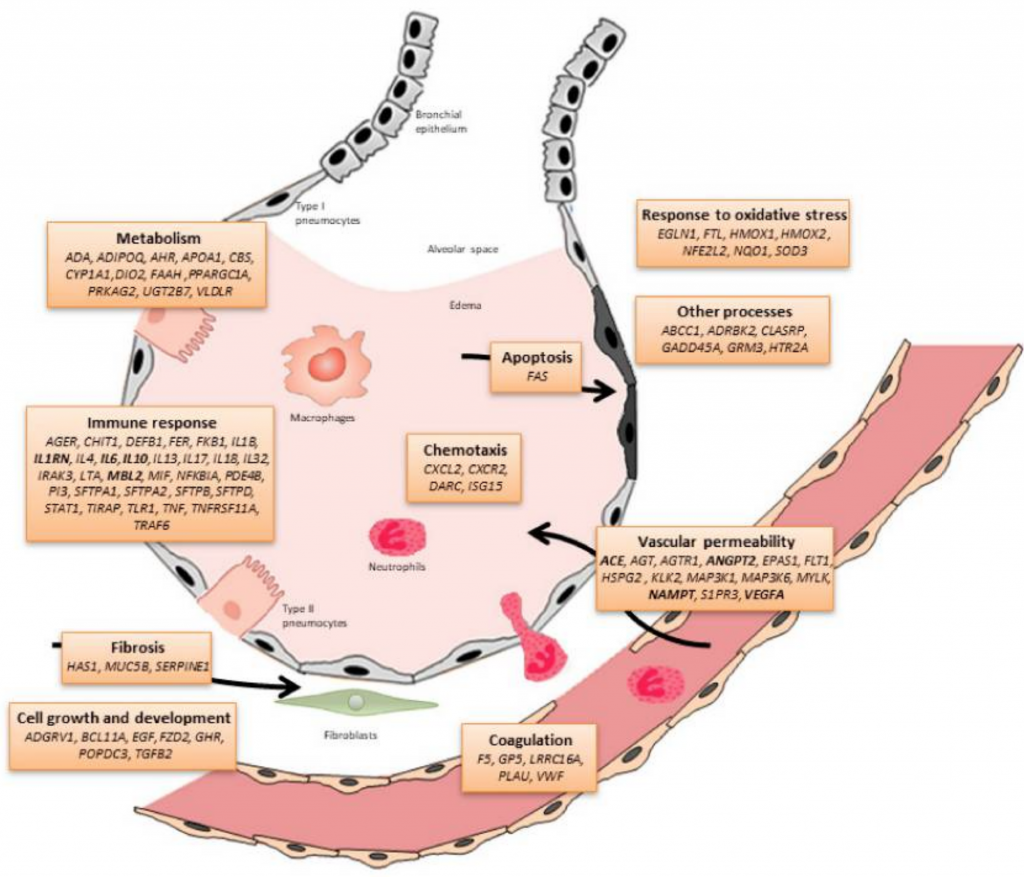
What are the factors behind leaking capillaries?
Vascular endothelial growth factor (VEGF) production occurs in the cells that make up blood vessels (endothelial cells). They cause increased blood vessels to form and also mediate the permeability, or leakiness, of the blood cells. So while VEGF can be a part of the problem with fluid building up around the lungs, it can also be part of the repair process when the injured lungs are healing.[ref]
Genetic variants in the VEGF gene can cause a person to create more or less VEGF during normal circumstances. These variants also have links to poor ARDS outcomes.
Plasma levels of angiopoietin-2 (ANG2) are linked to increased endothelial permeability, allowing fluid to move out of the capillaries. Animal models show that increasing ANG2 causes a disruption of the epithelial barrier and causes lung injury.[ref]
How are inflammatory markers involved?
TNF-alpha, a pro-inflammatory cytokine, is released for both acute infection and chronic inflammation. Levels of TNF-alpha increase with lung infections and COPD.[ref]
Interleukin-17 is another pro-inflammatory cytokine involved in amplifying the inflammatory response by recruiting more monocytes and neutrophils to an area. Higher IL-17 is linked to an increased risk of ARDS.[ref]
Another part of the innate immune system is the complement system, which is activated to enhance the body’s ability to clear out microbes as well as damaged cells. Mannose-binding lectin is one way of activating the complement system. Low levels of mannose-binding lectin, due to an MBL2 genetic variant, increase the risk for septic shock and ARDS.[ref][ref]
On the other hand, the overall picture of mannose-binding lectin and activation of the complement system isn’t entirely clear for the SARS coronavirus infections. Animal models here show that blocking the activation of the complement system lead to better outcomes for SARS coronavirus.[ref]
Related article:TNF-alpha: Inflammation and Your Genes
Oxidant-antioxidant balance:
The stress on the lung cells – whether from a mechanical insult such as a ventilator at the wrong setting or from bacterial or viral lungs – increases oxidative stress. One way that the body combats this is by increasing heme oxygenase-1, an enzyme that causes the reaction that converts heme (a pro-oxidant) into metabolites that are anti-inflammatory. Heme is part of the hemoglobin molecule and is vital for transporting oxygen. At the core of the heme molecule is iron, which is very reactive. So when the body increases heme oxygenase-1 to convert heme, it helps to balance out oxidative stress.[ref]
What does the future hold for ARDS?
Currently, medical professionals use therapy such as specific rates of ventilation and conservative fluid strategies to stop ARDS. The trials for pharmacological therapies are ongoing and target the inflammatory response. Additionally, trials using mesenchymal stem cells hold promise as well.[ref][ref][ref][ref][ref] There is hope for future treatments to reverse the course of ARDS.
How does COVID-19 cause ARDS?
For most people, COVID-19 infection will be asymptomatic or cause mild symptoms.
So what is the trigger for causing some people, mainly elderly people with comorbidities, to progress to ARDS?
Keep in mind that ARDS is a syndrome, so it is really a collection of symptoms and not a specifically defined disease.
First, let’s take a look at the co-morbidities, or underlying health conditions, linked to COVID-19, especially in the elderly, include:[ref]
- hypertension
- COPD
- diabetes
- cardiovascular disease
The virus causing COVID-19, now named SARS-CoV-2, enters cells in the body through a receptor called ACE2 (angiotensin-converting enzyme 2). Cells in the lower respiratory tract have ACE2 receptors on their surface. This allows the passage of the virus into the lung cells.[ref]
There are a number of different viral recognition receptors in the body that recognize the virus and then trigger MyD88 (below in the genetics section), which activates NF-κB, interferons, and several different inflammatory cytokines (also in the genetics section). This innate immune response needs to be regulated so that it doesn’t overwhelm the body. The infected lung cells are targeted, and if the initial infection isn’t cleared out, it can trigger “a series of immune responses and the production of cytokine storm in the body, which may be associated with the critical condition of COVID-19 patients.”[ref]
Initial data is showing that among COVID-19 patients that required ICU care, over 30% of them presented with ARDS.[ref]
Related article: Genetic variants linked to COVID-19 severity
So why are elderly people more prone to respiratory infections and pneumonia in general?
According to the statistics from the 2017-2018 flu season, between 61,000 and 80,00 people (mostly elderly) died of the flu in the United States. Worldwide, about 200 million cases of viral community-acquired pneumonia occur every year, with children and the elderly being hit hardest.[ref]
The elderly are hit hard by the flu and cold viruses due to frailty, an inability to clear mucous well, and a decline in the initial immune response. Senescence is the term applied to cells that no longer function and need to be cleared out of the body. Cellular senescence increases in older people, including senescence of immune cells (immunosenescence). This is one cause of the decrease in the ability of the immune system to clear out a viral infection quickly. “Infection is the primary cause of death in one-third of individuals aged 65 years and over.”[ref]
Related article: Genetic susceptibility to viruses
ARDS Genotype Report:
Not a member? Join here. Membership lets you see your data right in each article and also gives you access to the member’s only information in the Lifehacks sections.
Below are some of the genetic variants that research shows impacting either susceptibility to ARDS or mortality in ARDS. These variants have links to ARDS in general studies, not specific to COVID-19. Note that there are other genetic variants that influence ARDS (not found in 23andMe or AncestryDNA), so this is only giving you part of the genetic picture.
Increasing the permeability of blood vessels:
VEGFA gene: code for vascular endothelial growth factor, which both increases the permeability of blood vessels (leakiness) but is also important for recovery from ARDS with repair and growth of blood vessels
Check your genetic data for rs3025039 936C>T (23andMe v4, v5; AncestryDNA):
- C/C: typical (most common genotype)
- C/T: lower VEGF, increased risk of mortality in ARDS (~1.6x)
- T/T: lower VEGF, increased risk of mortality in ARDS (~4x)[ref]
Members: Your genotype for rs3025039 is —.
ANGPT2 gene: codes for ANG2, which increases the permeability (leakiness) of blood vessels
Check your genetic data for rs2442608 (23andMe v4; AncestryDNA):
- C/C: increased ANG2, slightly increase risk of ARDS in Caucasian populations[ref]
- C/T: typical risk
- T/T: typical
Members: Your genotype for rs2442608 is —.
Check your genetic data for rs2442630 (23andMe v5):
- G/G: slightly increased risk of ARDS in Caucasian pop.[ref]
- A/G: slightly increased risk of ARDS in Caucasians
- A/A: typical
Members: Your genotype for rs2442630 is —.
Check your genetic data for rs2515475 (23andMe v4):
- T/T: slightly increased risk of ARDS[ref]
- C/T: slightly increased risk of ARDS
- C/C: typical
Members: Your genotype for rs2515475 is —.
Immune response:
MBL2 gene: codes for mannose-binding lectin, which activates a part of the immune response. Low levels are generally linked with an increased risk of infections.
Check your genetic data for rs1800450 (23andMe v4, v5; AncestryDNA):
- C/C: typical
- C/T: lower mannose-binding lectin, increased risk of getting ARDS
- T/T: lower mannose-binding lectin, increased risk of getting ARDS[ref] (see explanation above, though, on lower MBL perhaps being beneficial in the SARS coronavirus)
Members: Your genotype for rs1800450 is —.
MyD88 gene: codes for a key activator of pro-inflammatory cytokines (NF-κB, TNF-alpha, and IL-6)
Check your genetic data for rs7744 (23andMe v4; AncestryDNA):
- A/A: typical
- A/G: decreased risk of ARDS in adults younger than 60
- G/G: decreased risk of ARDS in adults younger than 60[ref]
Members: Your genotype for rs7744 is —.
TNF gene: codes for TNF-alpha, an important inflammatory cytokine
Check your genetic data for rs1800629 308G>A (23andMe v4, v5; AncestryDNA):
- A/A: increased TNF-alpha, increased risk of ARDS in adults younger than 60[ref]
- A/G: increased TNF-alpha, increased risk of ARDS
- G/G: typical
Members: Your genotype for rs1800629 is —.
Note that a different study found a decreased risk of getting ARDS in children with sepsis with the rs1800629 A-allele[ref]. The increased TNF-alpha production perhaps may be a risk factor in adults, but not children or elderly patients. More studies are needed to know for sure.
IL17 gene: codes for interleukin-17, an inflammatory cytokine
Check your genetic data for rs2275913 (23andMe v4, v5; AncestryDNA):
- A/A: significantly decreased risk of ARDS (adjusted OR=0.28), decreased mortality rate[ref]
- A/G: decreased risk of ARDS, decreased mortality rate
- G/G: typical
Members: Your genotype for rs2275913 is —.
NAMPT gene: codes for a rate-limiting enzyme in the NAD+ salvage pathway. It acts as a modulator in the immune system.[ref]
Lifehacks:
The rest of this article is for Genetic Lifehacks members only. Consider joining today to see the rest of this article.
Access this content:
An active subscription is required to access this content.
Related Articles and Topics:
References: