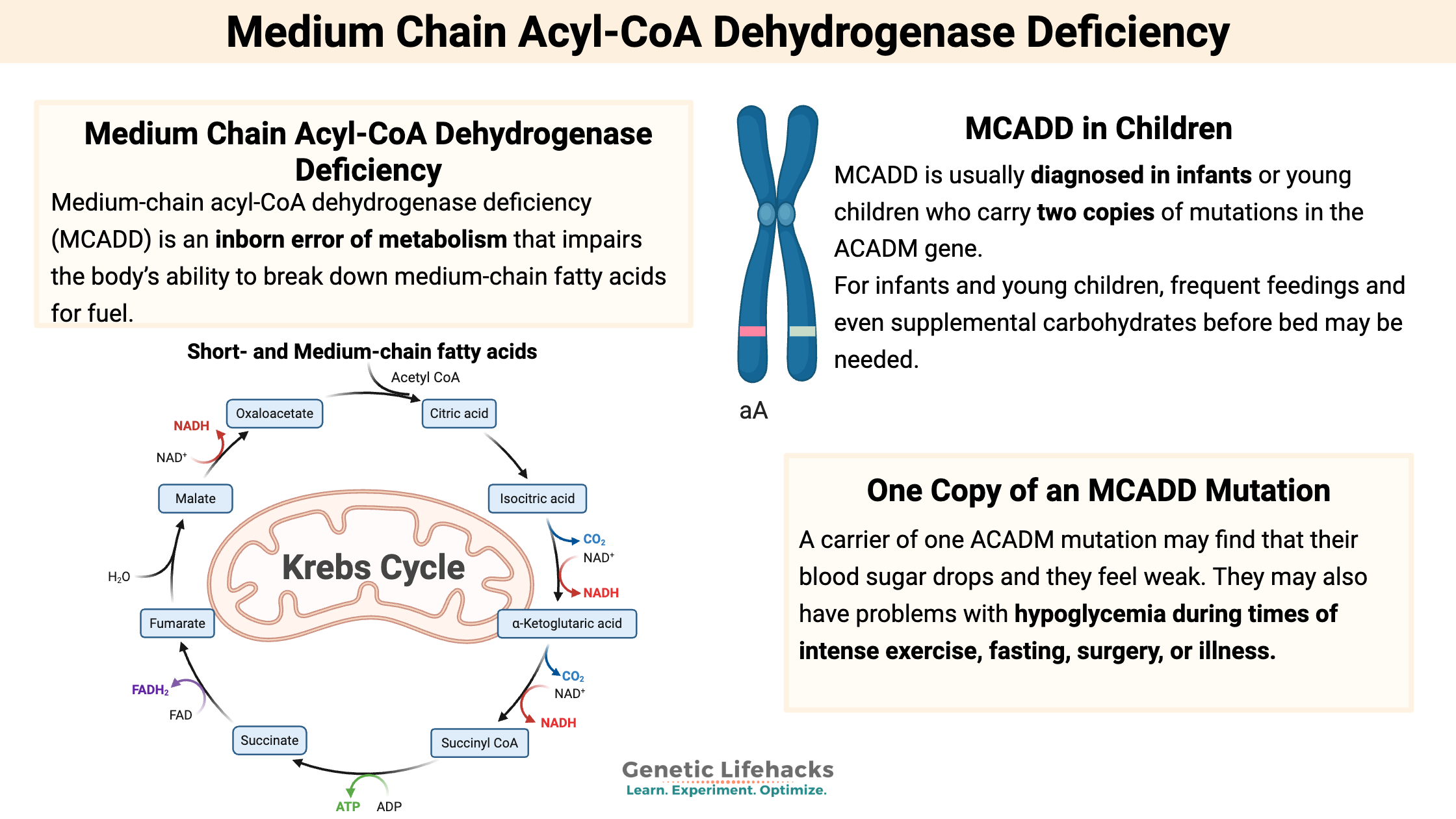
If you are a carrier (heterozygous) for one of the MCAD deficiency mutations, you may find that a diet that is higher in carbohydrates and protein, and lower in fat, may work better for you. In this era of carbs being demonized, MCADD carriers may need to buck the keto trend — or at least be alert for signs of hypoglycemia if eating a low-carb diet.
Coconut oil is really high in medium-chain fatty acids and is recommended to be avoided by people with MCADD.[ref]
Blois, B., et al. “Newborns with C8-Acylcarnitine Level over the 90th Centile Have an Increased Frequency of the Common MCAD 985A>G Mutation.” Journal of Inherited Metabolic Disease, vol. 28, no. 4, 2005, pp. 551–56. PubMed, https://doi.org/10.1007/s10545-005-0551-6.
Chang, Irene J., et al. “Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency.” GeneReviews®, edited by Margaret P. Adam et al., University of Washington, Seattle, 1993. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK1424/.
Christen, Matthias, et al. “ACADM Frameshift Variant in Cavalier King Charles Spaniels with Medium-Chain Acyl-CoA Dehydrogenase Deficiency.” Genes, vol. 13, no. 10, Oct. 2022, p. 1847. PubMed Central, https://doi.org/10.3390/genes13101847.
Dessein, Anne-Frédérique, et al. “A Novel Mutation of the ACADM Gene (c.145C>G) Associated with the Common c.985A>G Mutation on the Other ACADM Allele Causes Mild MCAD Deficiency: A Case Report.” Orphanet Journal of Rare Diseases, vol. 5, Oct. 2010, p. 26. PubMed, https://doi.org/10.1186/1750-1172-5-26.
Huidekoper, H. H., et al. “Prolonged Moderate-Intensity Exercise without and with L-Carnitine Supplementation in Patients with MCAD Deficiency.” Journal of Inherited Metabolic Disease, vol. 29, no. 5, Oct. 2006, pp. 631–36. PubMed, https://doi.org/10.1007/s10545-006-0355-3.
Huidekoper, Hidde H., et al. “Normal Rates of Whole-Body Fat Oxidation and Gluconeogenesis after Overnight Fasting and Moderate-Intensity Exercise in Patients with Medium-Chain Acyl-CoA Dehydrogenase Deficiency.” Journal of Inherited Metabolic Disease, vol. 36, no. 5, Sept. 2013, pp. 831–40. PubMed, https://doi.org/10.1007/s10545-012-9532-8.
Kennedy, Shelley, et al. “The First Three Years of Screening for Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCADD) by Newborn Screening Ontario.” BMC Pediatrics, vol. 10, no. 1, Nov. 2010, p. 82. Springer Link, https://doi.org/10.1186/1471-2431-10-82.
Lehotay, D. C., et al. “Blood Acylcarnitine Levels in Normal Newborns and Heterozygotes for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: A Relationship between Genotype and Biochemical Phenotype?” Journal of Inherited Metabolic Disease, vol. 27, no. 1, 2004, pp. 81–88. PubMed, https://doi.org/10.1023/B:BOLI.0000016636.79030.ad.
Madsen, K. L., et al. “Patients with Medium-Chain Acyl-Coenzyme a Dehydrogenase Deficiency Have Impaired Oxidation of Fat during Exercise but No Effect of L-Carnitine Supplementation.” The Journal of Clinical Endocrinology and Metabolism, vol. 98, no. 4, Apr. 2013, pp. 1667–75. PubMed, https://doi.org/10.1210/jc.2012-3791.
Mason, Emily, et al. “Medium‐chain Acyl‐COA Dehydrogenase Deficiency: Pathogenesis, Diagnosis, and Treatment.” Endocrinology, Diabetes & Metabolism, vol. 6, no. 1, Oct. 2022, p. e385. PubMed Central, https://doi.org/10.1002/edm2.385.
Randall, Morgan, et al. “Medium-Chain Acyl-CoA Dehydrogenase Deficiency in Adulthood: A Potential Diagnosis in a Patient with Mental Status Changes Suspected of Drug Toxicity.” Journal of Forensic Sciences, vol. 60, no. 4, July 2015, pp. 1101–03. PubMed, https://doi.org/10.1111/1556-4029.12808.
Sturm, Marga, et al. “Functional Effects of Different Medium-Chain Acyl-CoA Dehydrogenase Genotypes and Identification of Asymptomatic Variants.” PLoS ONE, edited by Steven R. Ellis, vol. 7, no. 9, Sept. 2012, p. e45110. DOI.org (Crossref), https://doi.org/10.1371/journal.pone.0045110.
VCV000003597.87 – ClinVar – NCBI. https://www.ncbi.nlm.nih.gov/clinvar/variation/3597/. Accessed 19 Jan. 2026.
Weiss, Katharina J., et al. “Free Carnitine Concentrations and Biochemical Parameters in Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Genotype-Phenotype Correlation.” Clinical Genetics, vol. 103, no. 6, June 2023, pp. 644–54. PubMed, https://doi.org/10.1111/cge.14316.
Zschocke, J., et al. “Molecular and Functional Characterisation of Mild MCAD Deficiency.” Human Genetics, vol. 108, no. 5, May 2001, pp. 404–08. PubMed, https://doi.org/10.1007/s004390100501.