Key takeaways:
- Short-chain Acyl-CoA Dehydrogenase Deficiency (SCADD) is a genetic disorder that affects the body’s ability to use short-chain fatty acids for energy.
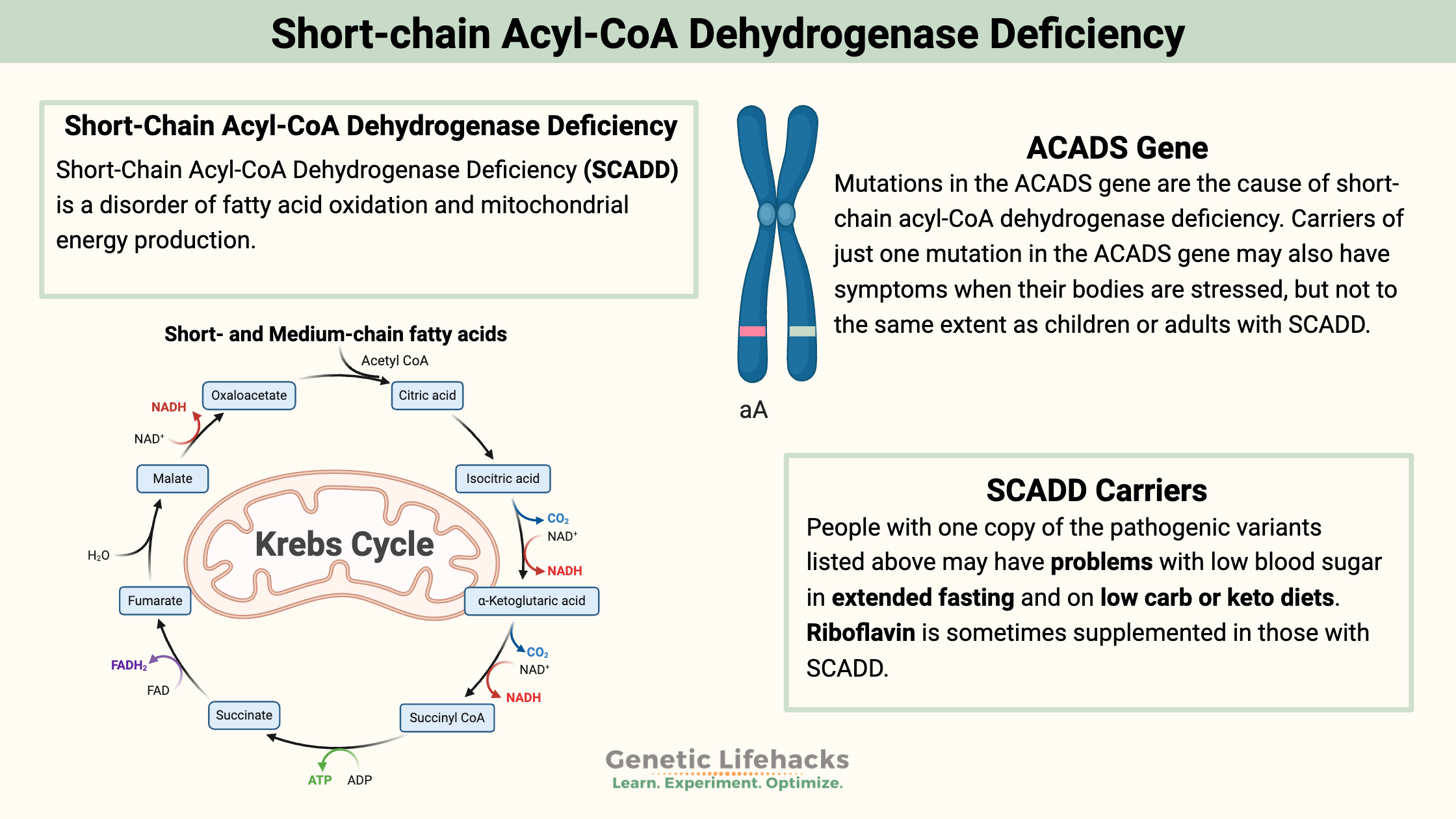
- Mutations in the ACADS gene cause this inborn error of metabolism.
- SCADD is an autosomal recessive disorder, meaning that two copies of the mutation are needed.
- Carriers of one copy of a mutation may have mild symptoms under stress and may struggle with a ketogenic diet or a prolonged fast.
Members will see their genotype report below, plus additional solutions in the Lifehacks section. Join today.
Using Fat for Fuel: Short-chain fatty acids and mitochondrial energy
The human body is wonderfully made and resilient enough to get energy from various food sources. Most people can survive and thrive using either carbs or fats. When our ancestors ran out of grain or potatoes, they could survive on fish or sheep. But certain genetic mutations can cause people not to burn fat for energy as efficiently.
Short-Chain Acyl-CoA Dehydrogenase Deficiency (SCADD) is a disorder of fatty acid oxidation and mitochondrial energy production. Think back to high school biology class when you learned that the mitochondria are the cellular “powerhouse”, making ATP or energy for our body. The ATP production process in the mitochondria can begin with either glucose (a sugar) or fatty acids.
If you start with glucose, glycolysis breaks down the glucose into two pyruvate molecules, which transform into acetyl-CoA. Fatty acids can also create acetyl-CoA, which begins the production of ATP in the Krebs cycle and then the electron energy transport chain.
Short-Chain Acyl-CoA Dehydrogenase enzyme deficiency
Short-Chain Acyl-CoA Dehydrogenase is an enzyme that converts short-chain fatty acids for use in the mitochondria. A deficiency of the enzyme makes it harder for an individual to use certain fatty acids for fuel.
Mutations in the ACAD gene can cause short-chain acyl-CoA dehydrogenase deficiency (SCADD). SCADD is classified as an inborn error of metabolism, which means it is an inherited disorder that affects a person’s ability to metabolize a specific nutrient. It is a rare disorder usually detected in infancy. Two copies of the mutation are needed to have the disorder.
Note that other fatty acid oxidation disorders include Medium-chain Acyl-CoA Dehydrogenase Deficiency and Long-chain Acyl-CoA Dehydrogenase Deficiency.
Symptoms:
Symptoms in infants with SCADD include hypoglycemia, lack of energy, vomiting, poor feeding, seizures, poor muscle tone, developmental delays, and failure to grow/thrive. This deficiency can be deadly if not managed with regular feedings.
Some newborn screenings now look for SCADD, and early detection and treatment can mitigate developmental problems.[ref] Increased screening has led to research showing that not all people who carry the SCADD mutations have symptoms.
Affected individuals may only have symptoms during times of fasting, illness, or other physiologic stress. During fasting or stress, the body can shift from using carbohydrates for energy to burning fatty acids. A recommendation for children with SCADD is to ensure they eat regularly to prevent hypoglycemia. (More info on SCADD)
Heterozygous carriers of ACADS mutations:
Carriers of just one mutation in the ACADS gene may also have symptoms when their bodies are stressed, but not to the same extent as children or adults with SCADD. Studies show varied results on this, so it may be that not everyone who is a carrier of an ACADS mutation has noticeable symptoms under physiological stress.[ref]
Situations in which symptoms of an ACADS mutation may be noticeable include a ketogenic diet, which causes cells to rely on fats for energy, or during illness or fasting. Endurance exercise or exercising while fasting also causes strain on mitochondrial energy production.
Recent research points to the mutations in ACADS causing the protein to misfold. It is theorized that SCADD symptoms may be due to decreased cellular energy combined with a toxic accumulation of the enzyme.[ref]
A cofactor in mitochondrial energy production is riboflavin (vitamin B2). Studies show that riboflavin deficiency can mimic SCADD in the effects on the heart (fibrosis and hypertrophy). The researchers found that increasing riboflavin caused an increase in the short-chain acyl-CoA dehydrogenase enzyme and helped to limit fibrosis.[ref]
Interestingly, there may be a benefit to lower ACADS expression. A study found that SCAD enzyme expression increases in aging in the liver, and that it could play a role in fatty liver disease. When the researchers partially knocked out the ACADS gene in mice, it prevented fatty liver.[ref]
ACADS: Genotype Report
Access this content:
An active subscription is required to access this content.
Lifehacks
The following changes may help with managing short-chain acyl-CoA deficiency carriers.
Avoiding Long Fasts:
People with one copy of the pathogenic variants listed above may have problems with low blood sugar during extended fasting. It is something to be aware of and plan for during fasting or illness. It may be especially true for kids.
Alcohol, fasted exercise:
Additionally, hard exercise without eating or drinking a lot of alcohol without eating may cause a drop in blood sugar levels.
Access this content:
An active subscription is required to access this content.
Related Articles and Topics:
Medium Chain Acyl-CoA Dehydrogenase Deficiency: Check your genetic data
Intermittent Fasting: Benefits from Changing Gene Expression
References:
An, Se Jin, et al. “Compound Heterozygous Mutations of ACADS Gene in Newborn with Short Chain Acyl-CoA Dehydrogenase Deficiency: Case Report and Literatures Review.” Korean Journal of Pediatrics, vol. 59, no. Suppl 1, Nov. 2016, pp. S45–48. PubMed Central, https://doi.org/10.3345/kjp.2016.59.11.S45.
Edhager, Anders V., et al. “Proteomic Investigation of Cultivated Fibroblasts from Patients with Mitochondrial Short-Chain Acyl-CoA Dehydrogenase Deficiency.” Molecular Genetics and Metabolism, vol. 111, no. 3, Mar. 2014, pp. 360–68. PubMed, https://doi.org/10.1016/j.ymgme.2014.01.007.
Ghosh, Sujoy, et al. “Short Chain Acyl-CoA Dehydrogenase Deficiency and Short-Term High-Fat Diet Perturb Mitochondrial Energy Metabolism and Transcriptional Control of Lipid-Handling in Liver.” Nutrition & Metabolism, vol. 13, Mar. 2016, p. 17. PubMed Central, https://doi.org/10.1186/s12986-016-0075-0.
Kruger, Claudia, et al. “Brain Transcriptional Responses to High-Fat Diet in Acads-Deficient Mice Reveal Energy Sensing Pathways.” PLoS ONE, vol. 7, no. 8, Aug. 2012, p. e41709. PubMed Central, https://doi.org/10.1371/journal.pone.0041709.
Nochi, Zahra, et al. “Short-Chain Acyl-CoA Dehydrogenase Deficiency: From Gene to Cell Pathology and Possible Disease Mechanisms.” Journal of Inherited Metabolic Disease, vol. 40, no. 5, Sept. 2017, pp. 641–55. PubMed, https://doi.org/10.1007/s10545-017-0047-1.
Rs121908006 RefSNP Report – DbSNP – NCBI. https://www.ncbi.nlm.nih.gov/snp/rs121908006. Accessed 13 Sept. 2022.
Tonin, Rodolfo, et al. “Clinical Relevance of Short-Chain Acyl-CoA Dehydrogenase (SCAD) Deficiency: Exploring the Role of New Variants Including the First SCAD-Disease-Causing Allele Carrying a Synonymous Mutation.” BBA Clinical, vol. 5, Mar. 2016, pp. 114–19. PubMed Central, https://doi.org/10.1016/j.bbacli.2016.03.004.
van Maldegem, Bianca T., Marinus Duran, Ronald J. A. Wanders, Hans R. Waterham, Tom J. de Koning, et al. “Fasting and Fat-Loading Tests Provide Pathophysiological Insight into Short-Chain Acyl-Coenzyme a Dehydrogenase Deficiency.” The Journal of Pediatrics, vol. 156, no. 1, Jan. 2010, pp. 121–27. PubMed, https://doi.org/10.1016/j.jpeds.2009.07.008.
van Maldegem, Bianca T., Marinus Duran, Ronald J. A. Wanders, Hans R. Waterham, and Frits A. Wijburg. “Flavin Adenine Dinucleotide Status and the Effects of High-Dose Riboflavin Treatment in Short-Chain Acyl-CoA Dehydrogenase Deficiency.” Pediatric Research, vol. 67, no. 3, Mar. 2010, pp. 304–08. PubMed, https://doi.org/10.1203/PDR.0b013e3181cbd57b.
Wolfe, Lynne, et al. “Short-Chain Acyl-CoA Dehydrogenase Deficiency.” GeneReviews®, edited by Margaret P. Adam et al., University of Washington, Seattle, 1993. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK63582/.