
Key takeaways:
- Mitochondrial energy production is essential for life, and mitochondrial dysfunction can negatively impact many aspects of health.
- Pyruvate dehydrogenase is at the heart of mitochondrial energy production.
- Mutations in the PDHA1 gene, which encodes pyruvate dehydrogenase, can cause mitochondrial dysfunction.
What is Pyruvate Dehydrogenase?
Pyruvate dehydrogenase is an enzyme that is essential for the production of cellular energy within the mitochondria. Acting as a catalyst, pyruvate dehydrogenase facilitates the conversion of pyruvate to acetyl-CoA, which is used in the citric acid (Krebs) cycle and ultimately leads to the production of ATP.
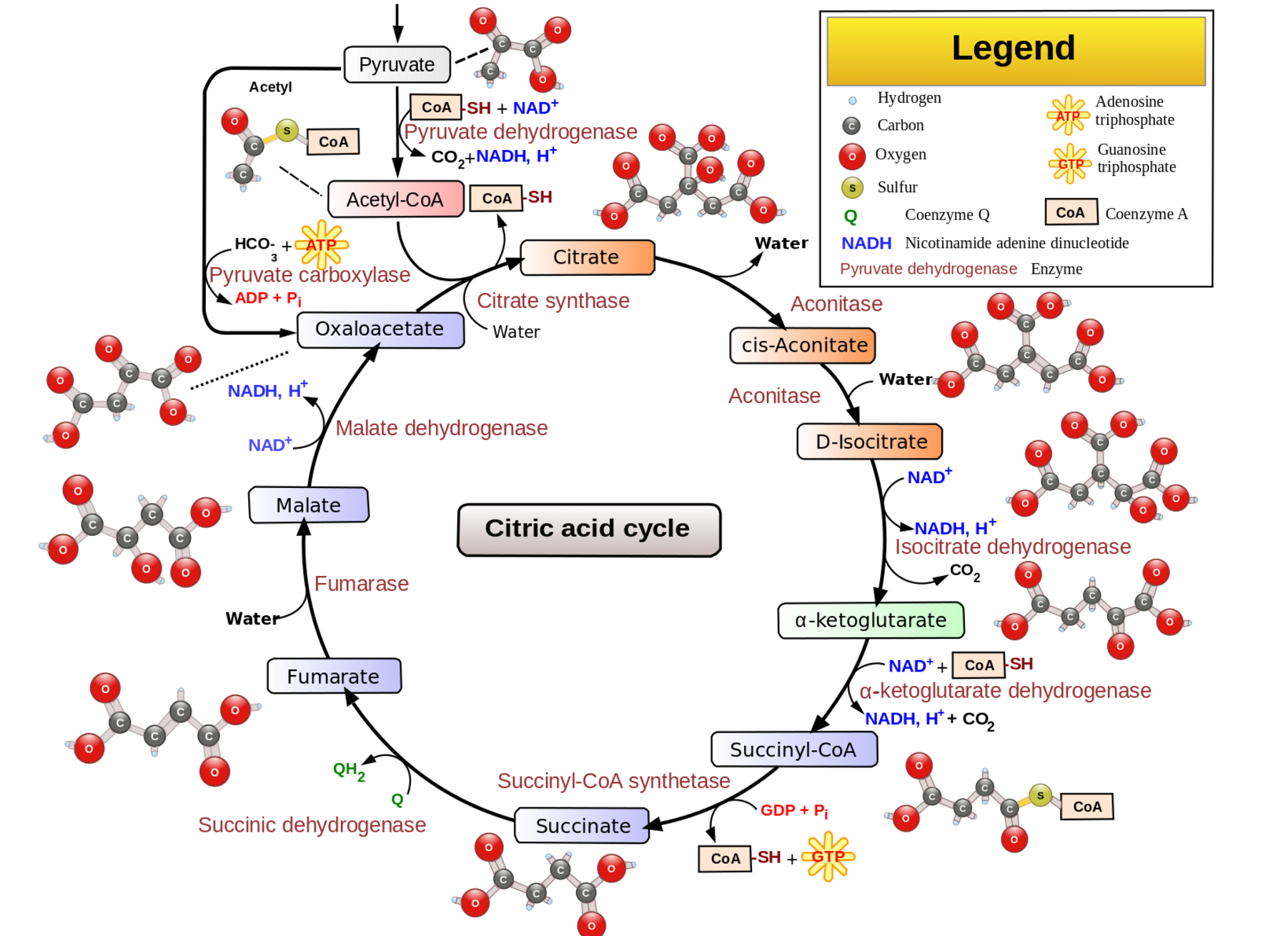
Given its essential function, deficiencies in pyruvate dehydrogenase can have severe implications for overall health and development.
Background science: Mitochondrial energy production
Mitochondria generate most of the ATP used by cells (yep, the powerhouse of the cell). While glycolysis in the cytosol also produces some ATP, mitochondria are the primary source.
Key concepts:
- ATP (adenosine triphosphate): ATP is the molecule used for energy in cellular processes in all life forms. The phosphate bonds in ATP are easily broken, releasing energy when adenosine triphosphate (ATP) becomes adenosine diphosphate (ADP).
- Citric Acid Cycle (Krebs cycle): In the mitochondria, the citric acid cycle converts pyruvate to acetyl-CoA, which is used to produce ATP.
- Oxidative phosphorylation: This process, located on in the inner membrane of the mitochondria, creates more ATP using products from the citric acid cycle.
With this in mind, let’s dive into why pyruvate metabolism is important.
Why is pyruvate important?
Pyruvate is produced from glucose during glycolysis –the breakdown of glucose for energy. Pyruvate is used by cells in several ways:
- used in the citric acid cycle to produce energy
- converted back to glucose via gluconeogenesis
- converted to lactic acid during fermentation or when oxygen is lacking
The reaction facilitated by pyruvate dehydrogenase is:
Pyruvate + CoA + NAD+ → Acetyl-CoA + CO2 + NADH
This reaction is vital for several reasons:
- Energy production: Acetyl-CoA enters the citric acid cycle, leading to the production of NADH and FADH₂, which are essential for ATP production via oxidative phosphorylation.
- Biosynthetic pathways: Acetyl-CoA also serves as a precursor for fatty acid synthesis, cholesterol synthesis, and the synthesis of other important biomolecules.
- Regulation of cell metabolism: The pyruvate dehydrogenase complex is a key regulatory point in cellular metabolism, integrating signals from various metabolic pathways to control the production of energy.
Pyruvate Dehydrogenase Deficiency:
What happens when pyruvate dehydrogenase is lacking?
Pyruvate dehydrogenase deficiency is a metabolic disorder caused by mutations in the PDHA1 gene or other genes related to the pyruvate dehydrogenase complex. It results in impaired conversion of pyruvate to acetyl-CoA. The mutations cause a buildup of lactate, which affects neurological and muscle development. [ref]
Infants born with two copies of mutations in the pyruvate dehydrogenase gene are diagnosed with pyruvate dehydrogenase deficiency in infancy. This is a very rare genetic disease that can be fatal in childhood.
Genetic basis:
Mutations in the PDHA1 gene, located on the X chromosome, are the primary cause of pyruvate dehydrogenase deficiency. Males, having only one X chromosome, are more severely affected.
In addition, other genes in the pyruvate dehydrogenase complex, such as PDHB or DLD, can cause pyruvate dehydrogenase deficiency.
In pyruvate dehydrogenase deficiency, there are several metabolic disruptions that occur:[ref]
- First, the decreased ability to use pyruvate in the citric acid cycle for ATP production causes both a decrease in mitochondrial energy production and an accumulation of pyruvate that then is shunted to lactate production.
- Pyruvate dehydrogenase deficiency can lead to neurological or muscular symptoms due to the cellular energy deficit. Neurological development is often impaired.
Other proteins that are part of the pyruvate dehydrogenase complex include dihydrolipoyl transacetylase (DLAT) and dihydrolipoyl dehydrogenase (DLD).[ref]
Beyond the severe genetic deficiency of pyruvate dehydrogenase, disruptions in this pathway may be implicated in conditions like ME/CFS and Long Covid.
A Link to ME/CFS and Long Covid?
Altered cellular energy production due to mitochondrial dysfunction is one theory behind conditions like Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) and Long Covid. [ref]
To be clear: Research does not show that people with ME/CFS have a mutation in the PHDA1 gene. However, impaired energy production along with excess lactate is a common thread.
A December 2016 study of metabolites in ME/CFS (Chronic Fatigue Syndrome) patients found indicators showing impaired pyruvate dehydrogenase. It concludes: “These findings are in agreement with the clinical disease presentation of ME/CFS, with inadequate ATP generation by oxidative phosphorylation and excessive lactate generation upon exertion.”[ref]
The researchers found that people with both moderate and severe ME/CFS had similar degrees of impaired bioenergetic function. They also had reduced glycolytic activity compared to healthy controls. The decrease in mitochondrial function is not due to deconditioning.[ref]
Studies also show increased lactate in the cerebrospinal fluid in ME/CFS.[ref] Elevated lactate levels correlate with the presence of post-exertional malaise (PEM).[ref] Long Covid patients have also had elevated lactate levels or altered lactate-to-pyruvate ratios in a couple of different studies.[ref][ref][ref]
Role in epigenetics:
Pyruvate dehydrogenase influences epigenetic modifications, specifically acetylation and lactylation, within the cell nucleus.
Turning on or off other genes:
Pyruvate dehydrogenase activity can also influence two epigenetic modifies – acetylation and lactylation. [ref] Acetylation and lactylation are ways that protein transcription can be regulated — turning on genes, essentially.
Lactylation, discovered in 2019, is when lactate directly acts in the cell nucleus to stimulate gene translation.[ref] When pyruvate dehydrogenase levels are throttled, there is increased conversion of pyruvate to lactate which can then impact gene expression.
The studies on lactylation are new and more research will likely further explain how lactate levels modify other proteins involved in cellular processes. For example, a 2024 mouse study shows that hypoxia, or low cellular oxygen, causes a mitochondrial shift due to lactylation of PDHA1.[ref]
What regulates the pyruvate dehydrogenase complex?
The pyruvate dehydrogenase complex (the PDHA1 gene plus DLD and DLAT) is primarily regulated by the phosphorylation of PDHA1, essentially adding a phosphate group to inactivate the enzyme. This is a regulatory mechanism that goes on all the time which skews carbohydrate metabolism toward cytoplasmic reduction of pyruvate to lactate under aerobic conditions.[ref]
Pyruvate is converted in the Krebs cycle (mitochondria) to acetyl-CoA, and this conversion is tightly regulated to prevent too much acetyl-CoA. So the amount of available acetyl-CoA acts as a feedback loop to suppress pyruvate dehydrogenase complex formation. Similarly, NADH is also a Kreb’s cycle product that inhibits the PDC.[ref]
Pyruvate Dehydrogenase: Genotype report
Access this content:
An active subscription is required to access this content.
Lifehacks:
For an infant diagnosed with pyruvate dehydrogenase deficiency, your doctor will recommend treatments appropriate for their individual case. Medications such as sodium dichloroacetate (DCA) or phenylbutyrate are often used to treat the genetic disease, depending on the mutation involved.[ref] Dietary interventions are also used.
Interestingly, sodium dichloroacetate (DCA) was shown in a study to help about half of patients with ME/CFS.[ref]
The following is based on recent research that may be of interest to someone who has high lactate levels and suspects that a partial impairment of pyruvate dehydrogenase is possible.
Dietary modifications:
Ketogenic diet:
A promising therapy for pyruvate dehydrogenase deficiency is a ketogenic diet. This reduces the need for glucose for energy and shifts towards using fatty acids. A study in children with pyruvate dehydrogenase deficiency found that a ketogenic diet “had a positive effect mainly in the areas of epilepsy, ataxia, sleep disturbance, speech/language development, social functioning, and frequency of hospitalizations.” Other studies also show promising results in children with pyruvate dehydrogenase deficiency.[ref][ref][ref]
Vitamins and Supplements:
Access this content:
An active subscription is required to access this content.
Related articles:
References: