Key takeaways:
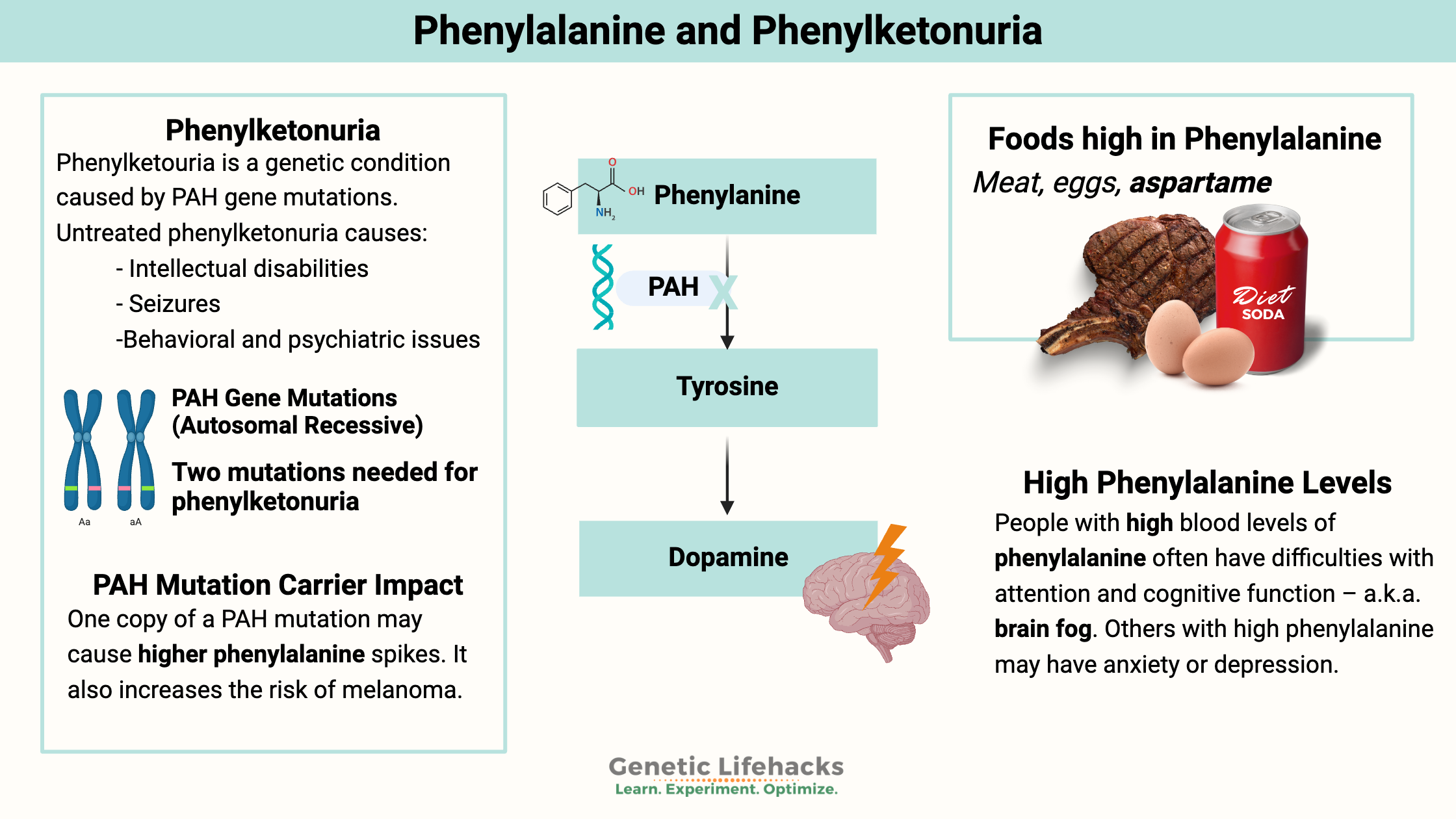
- Phenylketonuria, also called PKU, is a genetic metabolic disorder in which the amino acid phenylalanine is not metabolized correctly.
- PKU can cause intellectual disabilities, seizures, behavioral issues, and psychiatric illnesses if left untreated.
- It is one of the genetic diseases that infants are tested for when they are born.
Members will see their genotype report below and the solutions in the Lifehacks section. Consider joining today.
What is Phenylketonuria?
Phenylketonuria (PKU) is a hereditary condition caused by mutations in the PAH gene, which encodes the phenylalanine hydroxylase enzyme (PAH). The PAH enzyme breaks down excess phenylalanine, and the mutations reduce or eliminate enzyme function. As a result, dietary phenylalanine levels rise to potentially lethal levels.
Phenylketonuria is an autosomal recessive genetic disease, which means that the disease must be caused by mutations in both copies of the gene.
People who have only one copy of a defective gene are usually described as having no symptoms, but research does show that carriers may have more subtle changes. (More details on this below.)
What is phenylalanine?
Phenylalanine is an essential amino acid found in many foods containing protein. The PAH system in the liver metabolizes phenylalanine into tyrosine. Tetrahydrobiopterin (BH4), iron, and molecular oxygen are required for the hydroxylation of phenylalanine to tyrosine. Tyrosine is the precursor for the brain to make the neurotransmitter dopamine.
phenylalanine -> tyrosine -> dopamine
Which foods contain phenylalanine?
Eggs, chicken, beef, liver, milk, and soybeans contain phenylalanine. The artificial sweetener aspartame (NutraSweet, Equal, diet sodas) also breaks down into phenylalanine.
How does the body metabolize phenylalanine?
The main route of the metabolism of phenylalanine is in the liver. In the liver, the PAH (phenylalanine hydroxylase) enzyme converts phenylalanine into l-tyrosine in a reaction that uses tetrahydrobiopterin (BH4).
An alternate route of phenylalanine metabolism utilizes the aromatic L-amino acid decarboxylase enzyme (DDC gene). This enzyme (also known as DOPA decarboxylase) converts phenylalanine into PEA (phenethylamine), which is a trace amine that functions as a neuromodulator. This reaction uses vitamin B6 as a cofactor.
What happens to people with phenylketonuria?
Phenylketonuria (PKU) is caused by the excessive buildup of phenylalanine in the brain. Mutations in the PAH gene that cause the enzyme not to function are the main cause of phenylketonuria, but rare mutations that cause low BH4 levels are also linked to phenylketonuria.[ref]
The buildup of phenylalanine is a problem in infancy and causes neurological issues in the developing brain, resulting in intellectual disability.
About 1 in 10,000 infants are diagnosed with PKU, usually from infant blood spot testing. The use of infant testing allows for immediate dietary changes, eliminating most phenylalanine from the diet. A strict diet prevents the detrimental neurological effects on the developing brain.
Neuropsychological effects of PKU:
Even with dietary intervention, adults with PKU may have cognitive differences from the norm. One study found that adult PKU patients had altered processing speed, attention, inhibition, and motor control when compared to a control group.[ref][ref][ref]
Hemolysis in phenylketonuria
A recent study found that a buildup of phenylalanine can also cause an aggregation of proteins. In red blood cells, this may cause hemolysis or the breakdown of red blood cells.[ref]
Why is PKU a ‘common’ rare disease?
Phenylketonuria is the most common inborn error of amino acid metabolism in people of European descent. In some areas of Europe, more than 1% of the population carries one copy of a PAH mutation.[ref] On the other hand, PKU is much less common in many Asian countries.[ref]
Geneticists think that there must be some kind of advantage to carrying one copy of a PAH mutation. Without a ‘heterozygous advantage’, the percentage of people carrying the mutation statistically should be lower.
One possible reason for the frequency of PAH mutations in European and North African populations is that it may make pregnant women less susceptible to having miscarriages when exposed to the fungal toxin ochratoxin A.[ref]
Ochratoxin A is a mycotoxin created by certain types of mold that are commonly found on foods in humid areas, and exposure during pregnancy can cause miscarriages.[ref]
Phenylalanine is thought to act as a competitive inhibitor of ochratoxin A. Thus, researchers think that being resistant to the negative effects of this mycotoxin allowed more babies to be born with one copy of the mutation in areas where the fungal toxin has been prevalent.
Carrying one copy of a phenylketonuria mutation:
For an infant to be diagnosed with phenylketonuria, they usually need mutations in both copies of the PAH gene. Phenylketonuria is considered an autosomal recessive genetic disease.
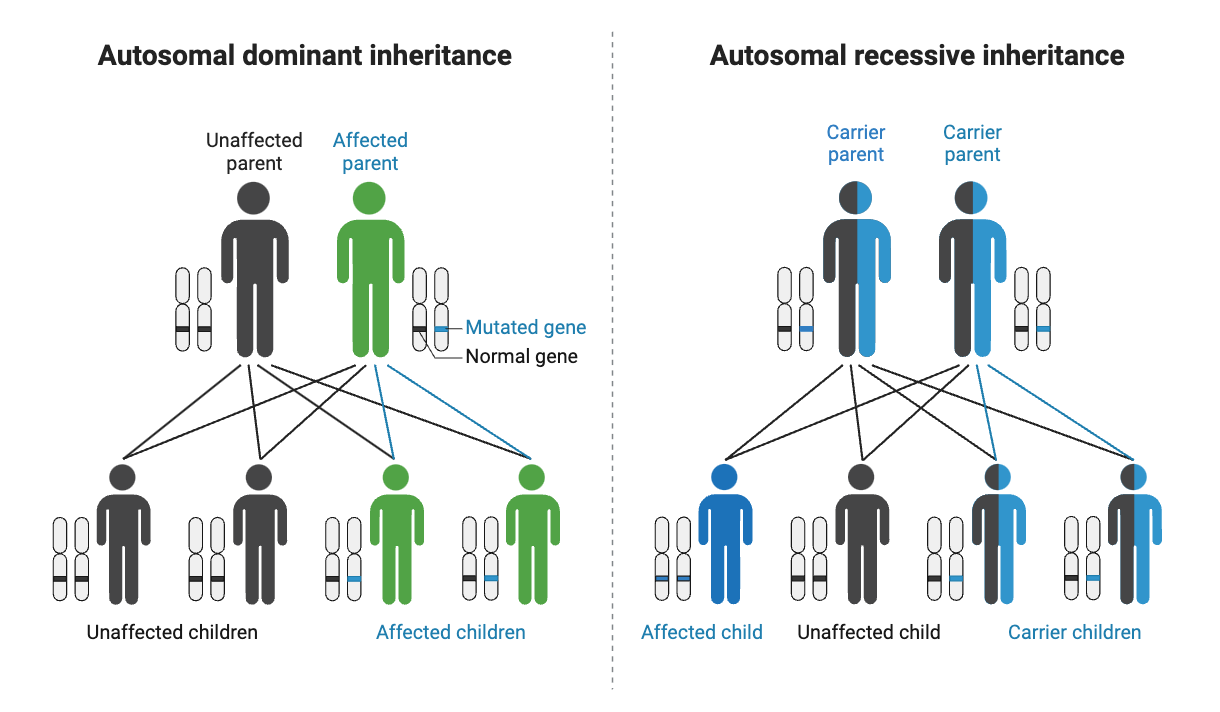
But what if you have one mutation? It is generally referred to as being a ‘carrier’ for phenylketonuria and is not associated with having the genetic disease.
Carriers of PAH mutations usually have less than 50% of the normal function of the PAH enzyme, but that is enough enzyme activity to prevent damage to the brain found in PKU.[ref]
With the advent of large-scale genetic testing, researchers now find that carrying one PAH mutation does cause some changes:
- People who carry one copy of a PAH mutation have a lower verbal recall, on average, when compared to a healthy control group. Additionally, they performed worse on divided attention and sensitivity to processing speed.[ref]
- In addition to being needed for dopamine, tyrosine is also a precursor for melanin. Carriers of phenylketonuria mutations are at double the normal risk of melanoma.[ref]
Phenylalanine levels in carriers of PAH mutation:
Eating a meal that contains a lot of phenylalanine (e.g., hamburger and milkshake) raises the concentration to a much higher level in heterozygous PAH mutation carriers.[ref] More recent testing using phenylalanine ‘challenge loads’ found a similar result, showing that carriers had a greater increase in phenylalanine blood levels.[ref]
Aspartame, which is 50% phenylalanine, raises phenylalanine levels in PAH mutation carriers more than in people without the mutation.[ref]
Interestingly, phenylalanine metabolism is lower than average in people with schizophrenia, although the research study didn’t find a statistical link to known PAH mutations. Lower phenylalanine metabolism likely indicates higher blood phenylalanine levels, depending on diet. BH4 (tetrahydrobiopterin) levels were also lower in schizophrenia patients than in the healthy control group.[ref]
High phenylalanine and brain fog:
People with high blood levels of phenylalanine often have difficulties with attention and cognitive function – a.k.a. brain fog. Others with high phenylalanine may have anxiety or depression. For adults with PKU, trials are underway with drugs that can substitute for the PAH enzyme.[ref]
Phenylketonuria Genotype Report:
Access this content:
An active subscription is required to access this content.
Related Articles and Topics:
Familial Mediterranean Fever: Mimics fibromyalgia, arthritis, inflammation
References:
Anand, Bibin G., et al. “Intrinsic Property of Phenylalanine to Trigger Protein Aggregation and Hemolysis Has a Direct Relevance to Phenylketonuria.” Scientific Reports, vol. 7, no. 1, Sept. 2017, p. 11146. www.nature.com, https://doi.org/10.1038/s41598-017-10911-z.
Andrade, Roseani, et al. “Phenylalanine and Tyrosine Metabolism Analysis in Heterozygotes for Phenylketonuria and in Healthy Individuals.” Journal of Inborn Errors of Metabolism and Screening, vol. 3, June 2019. SciELO, https://doi.org/10.1177/2326409815573962.
Arbesman, Joshua, et al. “Melanoma Cases Demonstrate Increased Carrier Frequency of Phenylketonuria/Hyperphenylalanemia Mutations.” Pigment Cell & Melanoma Research, vol. 31, no. 4, July 2018, pp. 529–33. PubMed Central, https://doi.org/10.1111/pcmr.12695.
Brown, Christine S., and Uta Lichter-Konecki. “Phenylketonuria (PKU): A Problem Solved?” Molecular Genetics and Metabolism Reports, vol. 6, Dec. 2015, pp. 8–12. PubMed Central, https://doi.org/10.1016/j.ymgmr.2015.12.004.
Feldmann, R., et al. “Neurocognitive Functioning in Adults with Phenylketonuria: Report of a 10-Year Follow-Up.” Molecular Genetics and Metabolism, vol. 126, no. 3, Mar. 2019, pp. 246–49. PubMed, https://doi.org/10.1016/j.ymgme.2018.12.011.
Hillert, Alicia, et al. “The Genetic Landscape and Epidemiology of Phenylketonuria.” The American Journal of Human Genetics, vol. 107, no. 2, Aug. 2020, pp. 234–50. ScienceDirect, https://doi.org/10.1016/j.ajhg.2020.06.006.
Kaufman, Seymour. “A Model of Human Phenylalanine Metabolism in Normal Subjects and in Phenylketonuric Patients.” Proceedings of the National Academy of Sciences of the United States of America, vol. 96, no. 6, Mar. 1999, pp. 3160–64. PubMed Central, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC15912/.
Kyei, Nicholas N. A., et al. “Maternal Mycotoxin Exposure and Adverse Pregnancy Outcomes: A Systematic Review.” Mycotoxin Research, vol. 36, no. 2, May 2020, pp. 243–55. Springer Link, https://doi.org/10.1007/s12550-019-00384-6.
Ledley, F. D., et al. “Molecular Analysis of the Inheritance of Phenylketonuria and Mild Hyperphenylalaninemia in Families with Both Disorders.” The New England Journal of Medicine, vol. 314, no. 20, May 1986, pp. 1276–80. PubMed, https://doi.org/10.1056/NEJM198605153142002.
Li, Nana, et al. “Analysis of the Genotype-Phenotype Correlation in Patients with Phenylketonuria in Mainland China.” Scientific Reports, vol. 8, no. 1, July 2018, p. 11251. www.nature.com, https://doi.org/10.1038/s41598-018-29640-y.
Moyle, J. J., et al. “Meta-Analysis of Neuropsychological Symptoms of Adolescents and Adults with PKU.” Neuropsychology Review, vol. 17, no. 2, June 2007, pp. 91–101. PubMed, https://doi.org/10.1007/s11065-007-9021-2.
Santangelo, Gabriella, et al. “Neuropsychological Profile in Parents of Adult Phenylketonuria Patients.” Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology, vol. 39, no. 1, Jan. 2018, pp. 161–64. PubMed, https://doi.org/10.1007/s10072-017-3181-5.
says, New Treatment for PhenylketonuriaClears Brain Fog |. PLOS Blogs Network. “New Treatment for Phenylketonuria (PKU) Clears Brain Fog.” DNA Science, 14 June 2018, https://dnascience.plos.org/2018/06/14/new-treatment-for-phenylketonuria-pku-clears-brain-fog/.
Teraishi, Toshiya, et al. “13C-Phenylalanine Breath Test and Serum Biopterin in Schizophrenia, Bipolar Disorder and Major Depressive Disorder.” Journal of Psychiatric Research, vol. 99, Apr. 2018, pp. 142–50. ScienceDirect, https://doi.org/10.1016/j.jpsychires.2018.01.019.
Weglage, J., et al. “Neurocognitive Functioning in Adults with Phenylketonuria: Results of a Long Term Study.” Molecular Genetics and Metabolism, vol. 110 Suppl, 2013, pp. S44-48. PubMed, https://doi.org/10.1016/j.ymgme.2013.08.013.
Debbie Moon is a biologist, engineer, author, and the founder of Genetic Lifehacks where she has helped thousands of members understand how to apply genetics to their diet, lifestyle, and health decisions. With more than 10 years of experience translating complex genetic research into practical health strategies, Debbie holds a BS in engineering from Colorado School of Mines and an MSc in biological sciences from Clemson University. She combines an engineering mindset with a biological systems approach to explain how genetic differences impact your optimal health.