
Key takeaways:
~ VLCADD is a genetic disorder caused by mutations in the ACADVL gene that impair the use of long-chain fatty acids for energy.
~ Symptoms and severity vary according to the mutation and can include hypoglycemia, heart problems, and muscle pain.
~ VLCADD is inherited in an autosomal recessive pattern, meaning that an individual generally needs two copies of the mutation to have the disease.
VLCADD: Inability to use long-chain fatty acids for energy
Very long chain acyl-CoA dehydrogenase deficiency, abbreviated VLCAD or VLCADD, is a genetic disorder that impairs the use of long-chain fatty acids for energy in the mitochondria. The disorder is caused by rare mutations in the ACADVL gene, which encodes the very-long-chain acyl-CoA dehydrogenase enzyme.
The VLCAD enzyme is important in mitochondrial energy production from fats and prevents the accumulation of harmful forms of fatty acids in the mitochondria.
A range of symptoms are seen in VLCADD, depending on the severity and the specific mutation. Symptoms can include low blood sugar (hypoglycemia), an enlarged heart, and muscle weakness. Most people with severe forms of VLCADD are identified as having it in infancy.
Let’s dig into the science of what’s happening, then look at your genes and possible solutions.
Fatty acid metabolism:
The mitochondria in your cells produce ATP, which is used for energy in all kinds of cellular reactions. Your mitochondria actually produce about 72kg of ATP a day, which is similar to an average body weight!
Within the mitochondria, a process called oxidative phosphorylation converts sugars, fats, and proteins into NADH and FADH2, which are then used as electron donors which drive the mechanisms that generate ATP. Oxidative phosphorylation (OXPHOS) occurs in the inner membrane of the mitochondria, where five complexes work together to produce protons that drive this process.
Fatty acids derived from fats we consume or that our bodies produce can be used for ATP production in mitochondria through fatty acid β-oxidation. They are especially important when the body has a need for more energy, such as when fasting, ill, or exercising.
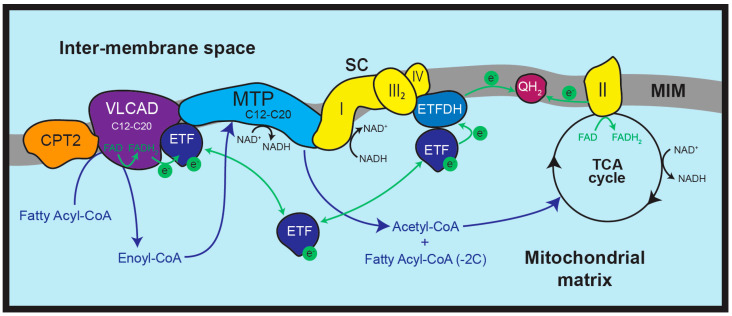
Fatty acids are chains of carbon atoms with hydrogen attached to them. They circulate in the body attached to albumin and then are taken into the cells when needed. First, an enzyme called acyl-CoA synthetase converts the fatty acid to a fatty acyl-CoA. Then, a second step involving carnitine converts it to a fatty acylcarnitine. The acylcarnitine is then transported across the inner mitochondrial membrane for use in OXPHOS.[ref]
Related article: Carnitine and mitochondrial transport
The first step for using fatty acids in oxidative phosphorylation is breaking them down within the inner membrane by dehydrogenase enzymes. There are three different dehydrogenase enzymes, and which one is used depends on the length of the fatty acid.
The acyl-CoA dehydrogenase enzymes include:
| Enzyme | Gene name | Fatty acid length |
|---|---|---|
| VLCAD (very-long-chain acyl-CoA dehydrogenase) | ACADVL | 12-20 carbons, C12 – C20 |
| MCAD (medium-chain acyl-CoA dehydrogenase ) | ACADM | C6-C12 |
| SCAD (short-chain acyl-CoA dehydrogenase) | ACADS | C4-C6 |
Related articles: SCADD and MCADD genes
The VLCAD enzyme is found on the inner membrane and converts long-chain fatty acids (12-20 carbons in length) into a form that can be used in mitochondrial energy production. Thus, mutations that significantly impair the function of the enzyme cause VLCADD.
The loss of VLCAD function not only decreases ATP production, but it also leaves an excess of the acylcarnitine form of the long-chain fat in the cell. This can then disrupt mitochondrial function, especially in muscle cells and heart cells, which require a lot of energy. [ref]
Forms of VLCADD and onset:
VLCAD deficiency (VLCADD) is more of a continuum or spectrum of phenotypes, rather than one specific disease. This is because the different mutations in the ACADVL gene can affect the function of the enzyme differently.
Early-onset, diagnosed in infancy (severe form):
Infant bloodspot screenings usually pick up the severe form of VLCADD, and these infant screenings have significantly reduced mortality from this condition. The severe form of VLCADD can cause cardiomyopathy, enlarged liver, rhabdomyolysis (muscle breakdown), and hypoglycemia. Before infant screening, if the baby was sick or not eating regularly, hypoglycemia could cause serious problems or even death.
With the advent of newborn screenings, research now shows the incidence of VLCADD to be about 1 in 9,300 infants.[ref]
Late-onset (mild):
Some people aren’t diagnosed with VLCADD until their late teens or adulthood. These cases often involve progressive limb weakness, exercise intolerance, rhabdomyolysis, and muscle pain. Biopsies can show moderate fat deposition in the muscles. Individuals with the milder forms of VLCADD usually have mutations that cause only a partial reduction of the enzyme’s function.[ref]
Inheritance of VLCADD:
VLCADD is an autosomal recessive condition, meaning that you need two mutations (one from each parent) to have it. More than 50 mutations have been identified in the ACADVL gene. Some mutations cause a partial failure of the enzyme, and others cause a completely non-functional enzyme. People with VLCADD often inherit two different mutations, which leads to combinations that can cause different phenotypes.
Carrier of one ACADVL mutation:
Most research studies that are looking at the severe form of VLCADD refer to the parents as “unaffected carriers”. However, under certain situations, the reduced ACADVL function can cause changes.
One case study involving a child with one pathogenic ACADVL mutation showed that it combined with common ACADVL variants to cause significantly reduced enzyme function.[ref] Another study in infants with a positive newborn screening for decreased VLCAD showed that they had a single copy of a mutation in the ACADVL gene. Further testing showed riboflavin deficiency, and supplemental riboflavin returned their VLCAD levels to normal. Interestingly, the mothers were on a vegan or vegetarian diet, which likely contributed significantly to the newborns having riboflavin deficiency.[ref]
A case study of a 17-year-old with rhabdomyolysis from exercise, as well as muscle pain and fatigue, was found to have one copy of a known ACADVL mutation along with other ACADVL variants that weren’t known to cause VLCADD.[ref]
Beyond mitochondrial energy:
Immune system impacts:
When monocytes and macrophages (white blood cells) are activated, the cells undergo metabolic changes, including an increase in long-chain fatty acid oxidation. In a study of people with long-chain fatty acid disorders, including VLCADD, researchers discovered that these individuals had slightly higher levels of inflammatory cytokines, including IL-6 and IL-1β.
Using macrophages from healthy people and macrophages with VLCAD, the researchers showed that VLCAD induces alternative macrophage activation in response to certain pathogenic signals. The changes due to VLCAD were subtle in effect.[ref] Other studies in people with long-chain fatty acid oxidation disorders also show a modest increase in inflammatory cytokine levels, including during exercise challenges.[ref]
Heart impacts:
People with VLCADD are at a higher risk of heart problems, such as cardiomyopathy. Arrhythmias and defects in the conduction of the cardiac signal are also common.[ref]
The heart muscle cells, called cardiomyocytes, preferentially use long-chain fatty acids to produce the large amount of ATP needed by the heart. In teens and young adults with VLCADD, there is a higher-than-normal risk of sudden cardiac arrest.[ref] Cell-line studies show that there’s an accumulation of long-chain acylcarnitine in the cardiomyocytes, which then alters the intracellular calcium concentration and interferes with signaling.[ref]
Genotype report:
Access this content:
An active subscription is required to access this content.
Lifehacks:
Treatment for VLCADD is set by the doctor depending on the severity of the disease. Generally, management includes dietary modifications (low-fat, high-carbohydrate), medium-chain triglyceride (MCT) oil, and L-carnitine supplementation.
Studies using gene therapy or mRNA are underway for patients with VLCADD, so keep an eye out for new discoveries and treatments if you or someone you know has VLCADD.[ref]
Diet:
Decreasing long-chain fatty acids in the diet is usually recommended for someone with two copies of the ACADVL mutations.
Sources of long-chain fats include:
- Nuts
- Seed oils, including canola, flax, chia, and sunflower
- Olive oil
- fatty fish
- Fatty meats
- Butter, full-fat dairy
Coconut oil is one fat that is mainly medium-chain fatty acids and may be a good substitute for other fats.
There’s no research showing that someone with one copy of an ACADVL mutation should limit long-chain fats. However, if you have symptoms associated with VLCADD, such as exercise intolerance or hypoglycemic episodes, it may be worth considering a lower-fat diet.
Supplements:
For people with adult-onset or mild VLCADD, clinicians often recommend supporting mitochondrial function.[ref] Supplements used in case studies include:
L-carnitine:
Carnitine is an essential part of moving fatty acids into the mitochondria, and supplemental carnitine may help someone who is low on carnitine or has genetic variants related to low carnitine levels.
Related article: Carnitine and your genes
Riboflavin:
Essential in the mitochondrial ATP production, riboflavin is a water-soluble B vitamin.
Related article: Riboflavin genes
CoQ10:
Within the mitochondrial inner membrane, CoQ10 plays a role in moving electrons for ATP production. This may be especially helpful for someone who has variants related to low CoQ10 or if on a statin, which can reduce CoQ10 levels.
Related article: CoQ10 genes
Resveratrol:
Cell-line studies show that resveratrol can help to mitigate the mitochondrial dysfunction shown in mild, but not severe, VLCADD.[ref][ref]
Recap of your genes:
| Gene | RS ID | Your Genotype | Effect Allele | Effect Allele Frequency | Notes About Effect Allele |
|---|---|---|---|---|---|
| ACADVL | rs113994167 | -- | C | 0.001 | likely to have two copies of a VLCADD pathogenic mutation, milder mutation |
| ACADVL | rs118204014 | -- | T | 0.0001 | likely to have two copies of a VLCADD pathogenic mutation |
| ACADVL | rs113994171 | -- | A | likely to have two copies of a VLCADD pathogenic mutation | |
| ACADVL | rs113994170 | -- | T | 0.00003 | likely to have two copies of a VLCADD pathogenic mutation |
| ACADVL | rs113994168 | -- | T | 0.00005 | likely to have two copies of a VLCADD pathogenic mutation |
| ACADVL | rs113690956 | -- | A | 0.00003 | likely to have two copies of a VLCADD pathogenic mutation |
| ACADVL | rs369560930 | -- | A | 0.00008 | likely to have two copies of a VLCADD pathogenic mutation |
| ACADVL | i5000095 | -- | C | 0.001 | likely to have two copies of a VLCADD pathogenic mutation, milder mutation |
| ACADVL | i5005484 | -- | A | 0.00003 | likely to have two copies of a VLCADD pathogenic mutation |
Related articles:
Medium Chain Acyl-CoA Dehydrogenase Deficiency: Check your genetic data
References: