
Key takeaways:
~ Medium-chain acyl-CoA dehydrogenase enzyme deficiency makes it hard to use medium-chain fats for energy.
~ MCAD deficiency is caused by two copies of mutations in the ACADM gene. It is usually discovered in infant blood screenings.
~ People with one copy of an ACADM mutation may have mild symptoms.
Members will see their genotype report below, plus additional solutions in the Lifehacks section. Consider joining today.
What is medium-chain acyl-CoA dehydrogenase deficiency?
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is an “inborn error of metabolism” that impairs the body’s ability to break down medium-chain fatty acids for fuel.
In a nutshell, the body can use either glucose (through glycolysis) or fatty acids (through beta-oxidation) as the basis for producing energy in the mitochondria in the form of ATP.
MCAD deficiency affects the body’s ability to efficiently use medium-chain fatty acids for energy.
MCAD deficiency in infants and children:
MCAD deficiency is a fairly rare genetic disorder, and it is usually only diagnosed in children who carry two copies of mutations in the ACADM gene (some are listed below).
Symptoms of MCADD generally occur when an infant or child hasn’t eaten, often due to being sick with a cold, ear infection, or the flu. Because the body can’t utilize fatty acids efficiently for energy, children with MCAD deficiency can’t switch to using fatty acids for energy efficiently. They will end up having severe hypoglycemia (low blood sugar), which can progress into a more serious metabolic crisis.[ref] It is thought that the onset of MCADD happens during an illness (viral, bacterial) because the immune system needs a lot of energy, while at the same time the child usually doesn’t feel like eating.
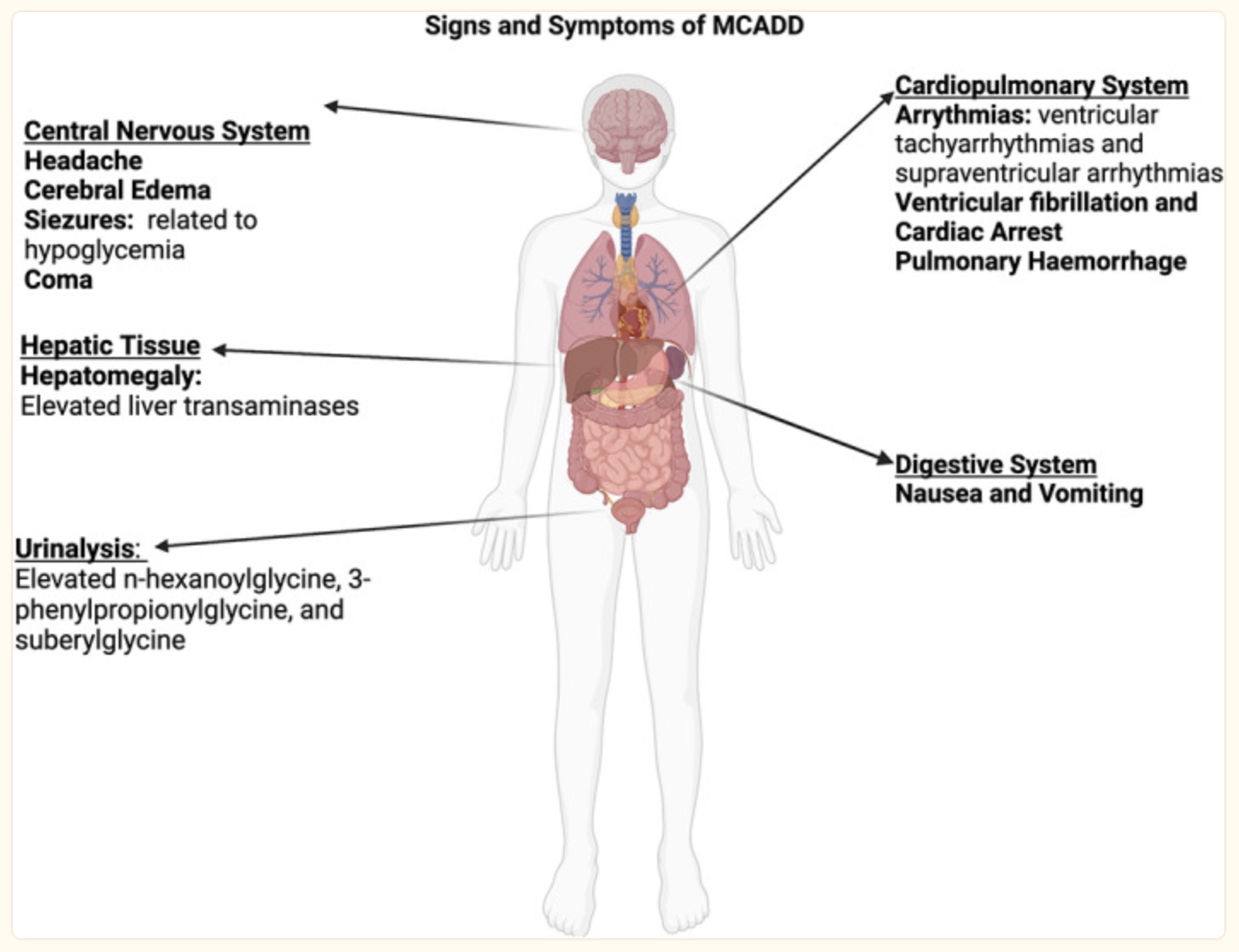
Not every child that has genetic mutations for MCAD deficiency ends up having problems with MCAD deficiency. Newborn screenings are now being done to identify infants with MCAD deficiency.[ref]
Under normal circumstances and while eating a regular diet, MCADD patients can compensate for the lack of beta-oxidation by using glycogen from the liver. However, some studies show that people with MCADD have overall impaired mitochondrial function in their skeletal muscles, which can result in muscle weakness.[ref]
Carnitine deficiency is a common occurrence in MCADD patients with more severe phenotypes and high concentrations of toxic acetylcarnitine.[ref]
Carrying one copy of an MCADD mutation:
It is now being recognized that people who carry one copy of an ACADM gene mutation may also have problems with hypoglycemia during times of intense exercise, fasting, surgery, or illness — basically, the times when your body may rely only on fatty acids instead of glucose for energy. People with one copy of the mutation may have no problems at all under normal conditions since fatty acid oxidation should still work — just at a less-than-optimal level.[ref][ref][ref][ref]
One study sums up: “As in other metabolic disorders, the distinction between “normal” and “disease” in MCAD deficiency is blurring into a spectrum of enzyme deficiency states caused by different mutations in the ACADM gene potentially influenced by factors affecting intracellular protein processing.”[ref]
Organic acids tests:
On an organic acids test, a common finding for someone with MCADD is that “medium-chain dicarboxylic acids are elevated with a characteristic pattern – hexanoylglycine (C6) > octanoylglycine (C8) > decanoylglycine (C10) – while ketones are inappropriately low.”[ref]
Interesting tidbit: King Charles Spaniel is a dog breed that is more likely to have mutations causing MCADD. One study showed that almost 1 in 10 King Charles Spaniels likely have at least a mild form.[ref]
MCADD Genotype Report:
Members: Log in to see your data below.
Not a member? Join here.
Why is this section is now only for members? Here’s why…
Lifehacks:
Nutrition and lifestyle considerations:
If you are a carrier (heterozygous) for one of the MCAD deficiency mutations, you may find that a diet that is higher in carbohydrates and protein, and lower in fat may work better for you. In this era of carbs being demonized, MCAD carriers may need to buck the keto trend — or at least be alert for signs of hypoglycemia if eating a low-carb diet.
Coconut oil is really high in medium-chain fatty acids and is recommended to avoid for people with MCADD.[ref]
Blood sugar drops:
The key is to keep an eye out for signs of low blood sugar. If you exercise hard, eating before a workout may be beneficial.
Anecdotally, drinking a lot of alcohol without eating anything can also cause hypoglycemia in people with one copy of the MCAD mutation.
Related Articles and Topics:
GLP-1 Receptor Agonists for Weight Loss: Genetic Interactions
GLP-1 receptor agonists, like semaglutide and liraglutide, are used for weight loss by increasing the body’s sensitivity to insulin and reducing hunger. However, genetic variants can alter the response in some people.
Short-chain Acyl-CoA Dehydrogenase Deficiency
If you have tried fasting or perhaps a ketogenic diet and felt horrible, there could be a genetic reason. You might carry a genetic mutation that causes SCADD (short-chain acyl-CoA dehydrogenase deficiency).
CYP2D6 gene and medication reactions
The CYP2D6 enzyme is responsible for metabolizing about 25% of commonly used medications. There are several fairly common genetic variants in CYP2D6 that affect how quickly you will break down a drug.
Magnesium deficiency? Genes that Impact Magnesium Levels
Are your magnesium levels low? Understanding your genes can help you decide whether you may need more magnesium in your diet or via supplements.