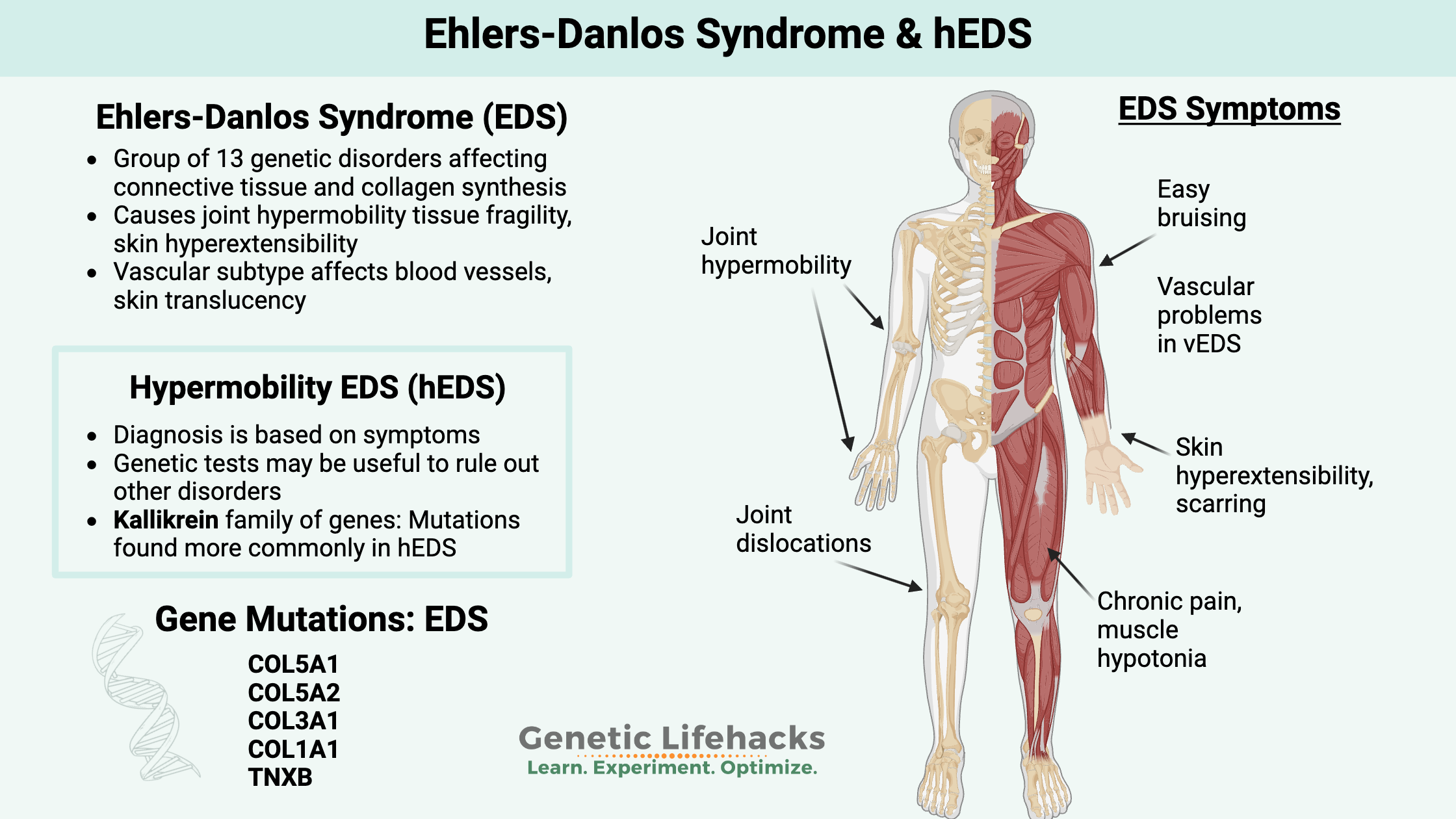
Key takeaways:
- Ehlers-Danlos Syndromes are a group of connective tissue disorders that change the way that collagen forms in the joints, ligaments, and skin.
- Rare genetic mutations in collagen genes cause changes to different connective tissues, giving rise to inherited EDS subtypes. Many, but not all, of these mutations are covered in 23andMe or AncestryDNA data (see the Genotype report section below).
- Hypermobility EDS (hEDS) is diagnosed based primarily on symptoms; genetic tests can be used to rule out other connective tissue disorders.
This article explores the research on Ehlers-Danlos syndrome and describes the genetic mutations that cause some of the disorder’s subtypes. You can learn more about how collagen disorders affect people and check your genetic data (23andMe v5 data works best).
Members will see their genotype report below, plus additional solutions in the Lifehacks section. Consider joining today.
Ehlers-Danlos Syndromes (EDS): Inherited connective tissue disorders and a hypermobility disorder
Ehlers-Danlos Syndromes (EDS) are a group of genetic disorders impacting the connective tissues in the body. It was first described in the early 1900s by Edvard Ehlers and Henri Danlos, two dermatologists, but it wasn’t formally classified until the 1940s. In 2017, the Ehlers-Danlos Syndrome International Consortium categorized 13 different subtypes and the criteria for diagnosis.
Most EDS subtypes are defined by the genetic mutation the person carries, with the exception of hypermobility EDS, which is diagnosed without genetic testing.
Symptoms of EDS vary by subtype. For example, classic EDS refers to people with joint hypermobility, skin hyperextensibility, easy bruising, tissue fragility, and scarring. Vascular EDS, caused by different genetic mutations, involves fragility of the tissue lining the blood vessels and internal organs.
This article covers:
- Background science: What goes wrong with the collagen in connective tissue
- Types of EDS: Which genes cause the different types of inherited EDS
- Hypermobility: The causes of hypermobility EDS (hEDS), plus the connection to mast cell activation and POTS
- Genotype report: How to check your genetic data for EDS mutations
- Lifehacks: Solutions based on research studies on hEDS and EDS
It’s a long article — bookmark it now.
Collagen and Connective Tissue: What goes wrong in EDS
EDS is a connective tissue disorder. Other connective tissue disorders include Marfan syndrome, lupus, rheumatoid arthritis, Churg-Strauss syndrome, and more.[ref]
What is connective tissue?
Between the bones, muscles, organs, and nerves is a connective tissue that holds everything together. Connective tissue is made up of collagen and elastic fibers, cells, and the extracellular matrix.
Tendons and ligaments are made up of dense connective tissue, with collagen fibers arranged in parallel for structure. Elastin is also found in the ligaments as well as in the skin.
Collagen is the most abundant protein in the body. The connective tissue cells, especially fibroblasts, secrete collagen into the extracellular matrix, forming a fibrous network.
Multiple types of collagen:
There are multiple types of collagen synthesized in different tissues. The different types of collagen can also interact with each other in forming different types of connective tissue.
- Type I: Most common type, found in skin, tendons, blood vessels, and bones.
- Type II: Cartilage, such as in your nose or ears
- Type III: Found supporting soft tissues
- Type IV: makes up part of the epithelium.
- Type V: Found in cell surfaces, hair, and the placenta.
The main amino acids that make up the collagen protein include glycine, proline, and alanine.
Synthesizing collagen:
Multiple genes encode the proteins important in collagen formation. Many of these start with the prefix COL, such as COL5A1. Interacting with the COL genes are specific enzymes needed in the formation of the connective tissue.
Inside connective tissue cells, a precursor form of collagen, called pro-collagen, is assembled. Then it is secreted into the extracellular space, and the final collagen fiber is assembled with the help of enzymes known as collagen peptidases.
The COL genes encode the proteins that form the procollagen, which is then acted upon by enzymes to create the different types of collagen in the body.
Most people diagnosed with classical Ehlers-Danlos syndrome (type III) have a single mutation in the COL5A1 or COL5A2 gene, which encodes proteins important in the formation of Type V collagen. Type I collagen and type V collagen work together to form collagen in certain areas of the body. You can’t live without type V collagen, and a complete lack of it is lethal in embryo formation.[ref]
In wound healing and skin thickness, type V collagen is important. One way that researchers have determined this is through animal studies. Animal models of EDS were created using mice that were bred to lack COL5A1. The lack of COL5A1 causes thin skin along with spontaneous wounds that don’t heal correctly.[ref]
Type V collagen is also important for the joints and spine. Certain mutations in COL5A1 are linked to progressive kyphoscoliosis, which is a curve in the upper back that is both sideways and an arched hump.[ref]
Symptoms of classical Ehlers-Danlos Syndrome:
A recent study involving 75 people with genetic confirmation of classical EDS illustrated the wide range of symptoms. The study participants had a mutation in the COL5A1 gene or the COL5A2 gene.
Symptoms reported for classical EDS include[ref]:
|
|
Facial features typical for EDS included epicanthal folds (upper eyelid skin fold), drooping eyelids, prominent eyes, eyes that are closer together than typical, low-set ears, and elongation in the area between nose and mouth.
Key point here:
Not everyone with a classical EDS mutation will have the same features and symptoms. There is a wide range of phenotypes, depending on the specific mutation as well as other genetic variants and environmental factors. Some will have more severe symptoms, while others will have milder issues.[ref]
Types of Ehlers-Danlos Syndrome:
Let’s dive into some details on the specific types of EDS.
Classical EDS:
Classic EDS, or EDS type I/II, refers to people with joint hypermobility, skin hyperextensibility, easy bruising, tissue fragility, and scarring. In addition to skin and joint issues, some people with classic EDS will also have vascular (blood vessel) complications.
Genetic mutations in classic EDS:
People with classic EDS have mutations in the COL5A1 or COL5A2 gene. A partial list of mutations in these genes is below in the Genotype report section. According to research studies, classical EDS is found in about 1 in 20,000 people.[ref]
Vascular Ehlers-Danlos Syndrome:
Considered a rare and severe disorder, vascular Ehlers-Danlos is characterized by fragile, thin skin and blood vessels. The endothelial tissue that lines the blood vessels, intestines, and uterus is prone to easily rupturing. Gastrointestinal tract complications are possible. This can result in an aneurysm, intestinal fistula, or bowel rupture. Usually diagnosed in their 20s or 30s, people with vEDS often have characteristic thin, translucent skin and easy bruising. Diagnosis often follows a major vascular event (e.g., stent, aneurysm), abdominal aneurysm, or bowel rupture.[ref][ref][ref]
Genetic mutations in vascular EDS:
Diagnosis with vascular Ehlers-Danlos includes a positive genetic test for a pathogenic mutation in the COL3A1 gene. While many rare genetic diseases require two pathogenic mutations for the disease to occur, vascular Ehlers-Danlos can sometimes be caused by carrying just one mutation in the gene.
There are over 500 known mutations in the COL3A1 gene, and the overall frequency of vEDS is 1 in 150,000 people.[ref]
Dermatosparaxis EDS:
Dermatosparaxis EDS, or EDS type VIIC, involves extremely fragile and lax skin. People with this subtype of EDS are often shorter in stature and have a blue discoloration of the whites of their eyes.
Genetic mutations in dermatosparaxis EDS:
Rare mutations in the ADAMS2 and ADAMS12 genes are responsible for dermatosparaxis EDS. These genes are involved in cleaving the collagen proteins at the right spot for them to be activated.[ref]
Periodontal Ehlers-Danlos Syndrome:
Another subtype of Ehlers-Danlos involves the gums and teeth. Periodontitis and tooth-root absorption are hallmarks of this subtype.[ref]
Genetic mutations in periodontal EDS:
The genetic component of periodontal EDS involves immune system genes that code for part of the complement system. People with periodontal EDS have a mutation in the C1R (complement 1 receptor) gene. In addition to severe periodontitis, people with periodontal EDS can also have easy bruising and joint hypermobility.[ref]
Classification of EDS Subtypes:
Here is a table that explains which genes are associated with the 13 types of EDS:[ref]
| Subtypes | Notable features | Gene(s) involved | Structural effect |
|---|---|---|---|
| Classical EDS | Skin fragility, hyperextensibility, scarring Joint hypermobility, dislocations, easy bruising, and hernia |
COL5A1 COL5A2 Rarely COL1A1 |
Affects the primary structure and processing of collagen |
| Classical-like EDS | Skin fragility, bruising, hyperextensibility Joint hypermobility, dislocations, mild muscle weakness, atrophy, neuropathy, edema, organ prolapse |
TNXB | Affects myomatrix structure and function |
| Cardiac-valvular EDS | Joint hypermobility, Skin hyperextensibility, Severe defects of cardiac valves | COL1A2 | Affects the primary structure and processing of collagen |
| Vascular EDS | Aneurysm, rupture of arteries, Perforation of gastrointestinal organs, Rupture of the uterus, Skin translucency, bruising | COL3A1 Rarely COL1A1 |
Affects the primary structure and processing of collagen |
| Hypermobile EDS (most common type) |
Joint hypermobility, Velvety/soft skin. Slightly hyperextensible skin, Common joint dislocations, Chronic pain | Causative gene unidentified |
|
| Arthrochalasia EDS | Severe joint hypermobility at birth, dislocation of bilateral hips, Skin hyperextensibility | COL1A1 COL1A2 |
Affects the primary structure and processing of collagen |
| Dermatosparaxis EDS | Extreme skin fragility and laxity, Blue discoloration of the sclera, Short fingers and stature | ADAMTS2 | Affects the primary structure and processing of collagen |
| Kyphoscoliotic EDS | Kyphoscoliosis (spinal curvature), Joint hyperflexibility, Muscle hypotonia, Ocular fragility, Skin hyperextensibility | PLOD1 FKBP14 |
Affects the folding and cross-linking of collagen |
| Brittle cornea Syndrome | Fragile cornea with risk of rupture, Blue sclerae, Early keratoconus, Severe myopia, Detachment of retina, Deafness | ZNF469 PRDM5 |
Affects intracellular processes |
| Spondylodysplastic EDS | Hypotonia of muscles, Short stature and bowing of limbs, Delayed cognitive and motor development | B4GALT7 B3GALT6 SLC39A13 |
Affects the biosynthesis of glycosaminoglycan and intracellular processes |
| Musculocontractural EDS | Multiple contractures, Club foot, Early craniofacial abnormalities, Skin hyperextensibility | CHST14 DSE |
Affects the biosynthesis of glycosaminoglycans |
| Myopathic EDS | Congenital muscle weakness and atrophy, Contractures of proximal joints, Joint hypermobility | COL12A1 | Affects myomatrix structure and function |
| Periodontal EDS | Early, severe gum disease, Detachment of gingiva, Pretibial plaques, Easy bruising, hypermobility | C1R, C1S | Affects complement pathways |
Hypermobile Ehlers-Danlos Syndrome (hEDS):
Hypermobility (or Hypermobile) Ehlers-Danlos syndrome (hEDS), or EDS type III, is characterized by laxity in the ligaments. This can cause joint dislocation, joint weakness, joint pain, nerve pain, or muscle pain. Additionally, some people can have an increased risk of early-onset arthritis and osteoporosis.[ref][ref]
Hypermobile Ehlers-Danlos is currently diagnosed based on symptoms. The hypermobility of joints in hEDS is generally noticeable in early childhood. In other words, hEDS isn’t something that develops as an adult, but instead, the hypermobility aspects are apparent from early childhood.[ref]
With an hEDS diagnosis, genetic testing is used to rule out other connective tissue disorders, including classic EDS mutations, Marfan syndrome, Loeys-Dietz syndrome, osteogenesis imperfecta, and lateral meningocele syndrome.[ref][ref]
Hypermobility Spectrum Disorder (HSD):
Similar to hEDS, hypermobility spectrum disorder (HSD) is a term being applied to the group of connective tissue disorders that involve hypermobility in one or more joints, subluxations, and joint dislocations.[ref][ref] Joint hypermobility syndrome is another term given when diagnosing people with joint hypermobility and no defined molecular or genetic basis.[ref]
Prevalence of diagnosis:
In the UK, a recent study found that about 1 in 500 people were diagnosed with either joint hypermobility or Ehlers-Danlos Syndrome. This shows a much greater prevalence than earlier diagnoses of EDS based on genetic testing (1 in 20,000) or even the previous prevalence of hEDS (1 in 5,000).[ref]
When you read through the diagnostic criteria for hEDS or hypermobility spectrum disorder, it seems like this is cut-and-dry. Certain symptoms = certain diagnoses. But the reality is that everyone is unique, and not everyone fits into a specific box.[ref]
Genetics studies on hEDS:
Looking at the genetic susceptibility for hEDS can give us a better picture of what is going on in the body.
Some individuals have rare EDS mutations that weren’t initially identified:
A 2023 whole-genome study examined people with hEDS symptoms but without a genetic diagnosis. The researchers found that out of 174 patients, there were 10 with EDS mutations found on the whole genome testing. Additionally, there were multiple other mutations that were likely related to the EDS symptoms in other hEDS patients.[ref]
Neuroimmune and Connective tissue pathways: A 2025 genome-wide association preprint study from MUSC looked at over 1,800 cases of hEDS along with thousands of ancestry-matched controls. The two top genetic hits showed that variants in the ACKR3 gene and the SLC39A13 gene were likely involved. ACKR3 ties in pain signaling and neuroimmune pathways, while SLC39A13 is involved in connective tissue development.[ref]
Collagen synthesis mutations:
Another study involving 100 patients with hEDS found that 1 out of the 100 patients had a mutation in the MIA3 gene, which encodes a protein involved in collagen synthesis.[ref] There are multiple genes involved in the formation and breakdown of the extracellular matrix, so some people with hEDS may have rare mutations in different genes that affect joint laxity.
Kallikrein mutations identified in hEDS:
The kallikrein family of enzymes breaks apart protein bonds. They are a class of protein-degrading enzymes. The body is constantly making new proteins and breaking down proteins to alter or inactivate them. While we often focus on the creation of proteins from genes, the degradation and recycling of proteins are equally important and balance everything out. Humans have 15 genes in the kallikrein family, and the enzymes interact with the extracellular matrix and connective tissue.[ref]
A 2024 preprint study (Gensemer et. al.) investigated the prevalence of rare variants in families with hEDS. The genetic results pointed the researchers to look at kallikrein, discovering a rare mutation in the KLK15 (kallikrein 15) gene. They then used mice with the KLK15 gene knocked out to see if deleting the gene caused symptoms similar to hEDS. The researchers found that the lack of KLK15 caused changes in the tendons of the mice and also caused valve dysfunction in the hearts of the mutant mice. Taking it another step, the researchers looked at genetic data for 197 hEDS patients and found that about of third of them had a rare mutation in one of the KLK genes.[ref]
Interestingly (for hEDS), kallikrein interacts with the immune response and blood pressure. This ties into the alterations in blood pressure seen in some people with hEDS – as well as the immune activation and mast cell activation. Kallikreins can be found in plasma or in tissue. Plasma kallikrein is involved in blood pressure regulation through interaction with bradykinin, and it is one way that plasminogen can be activated to plasmin in forming clots. Tissue kallikreins are also involved in blood pressure via the kidneys and are involved in healthy arteries and heart function. In the immune system, tissue kallikrein (KLK1) increases significantly in the lungs during an influenza infection and modulates inflammation. KLK5 and KLK12 interact with coronavirus infections.[ref][ref][ref][ref]
Related articles: Plasminogen and PAI1, fibrinogen
Does having flexible joints mean that you have Ehlers-Danlos or hEDS?
Up to 25% of kids and teens are considered to have joint hypermobility. Girls are more likely than boys to exhibit this tendency, and it is thought to cause no complications in kids. Thus, simply being a flexible teenage girl does not necessarily mean it is EDS… instead, other symptoms also need to be present, such as skin laxity, easy bruising (without another cause), or joint dislocations.[ref]
Along with joint hypermobility, many with hypermobility EDS diagnosis also have other co-morbidities.
Commonly found together: POTS, MCAS, Long Covid, Fibromyalgia, and hEDS
A pattern emerges showing that people with hEDS often also have other conditions at a much higher rate than normal. The trifecta for many is POTS, MCAS, and hEDS, but there is also a statistical overlap with long Covid and fibromyalgia.
POTS and hEDS:
POTS stands for postural orthostatic tachycardia syndrome. It is defined as an increase in heart rate of 30 beats/minute (adults) or 40 beats/minute (kids, teens) when going from lying down to standing up.
There is an overlap between people with POTS and hEDS that is not well explained.
Some estimates show that over 40% of people with hEDS also have POTS, but not all research agrees. Part of the difference in studies is that POTS can be defined in different ways in different clinics. Additionally, much of the data in the studies is self-reported rather than measured in a clinic.[ref]
A recent study at Penn State investigated the link between POTS and hEDS. The study included 91 people with clinically diagnosed POTS. Of those, 31% met the clinical criteria for hEDS. Notably, an additional 24% of participants had some joint hypermobility without meeting the hEDS criteria.[ref]
Related article: Genetic variants linked to POTS
Mast Cell Activation Syndrome and hEDS:
Similar to the overlap between POTS and hEDS, there is an unusually high number of people diagnosed with hEDS along with mast cell activation syndrome (MCAS). Some people have all three diagnoses – POTS, hEDS, and MCAS.
A recent study showed the overlap between MCAS, POTS, and hEDS diagnoses in medical records was 31%.[ref]
Is it totally clear that there is a link between MCAS, POTS, and hEDS?
Well… no. MCAS and hEDS have newly evolving clinical criteria and changing definitions. POTS is often diagnosed without a clinical test. There is murkiness here in the research studies.[ref]
Murky questions acknowledged, let’s take a look at what could be going on.
A biological basis for the connection between MCAS, POTS, and hEDS:
Mast cells are part of the immune system, reacting as part of the body’s first line of defense against invaders. When a mast cell is activated, such as in response to an allergen or viral particle, it will degranulate and release mediators such as histamine, heparin, tryptase, and/or serotonin. Additionally, mast cells can release cytokines such as TNF-alpha or IL-4 in response to specific activators.
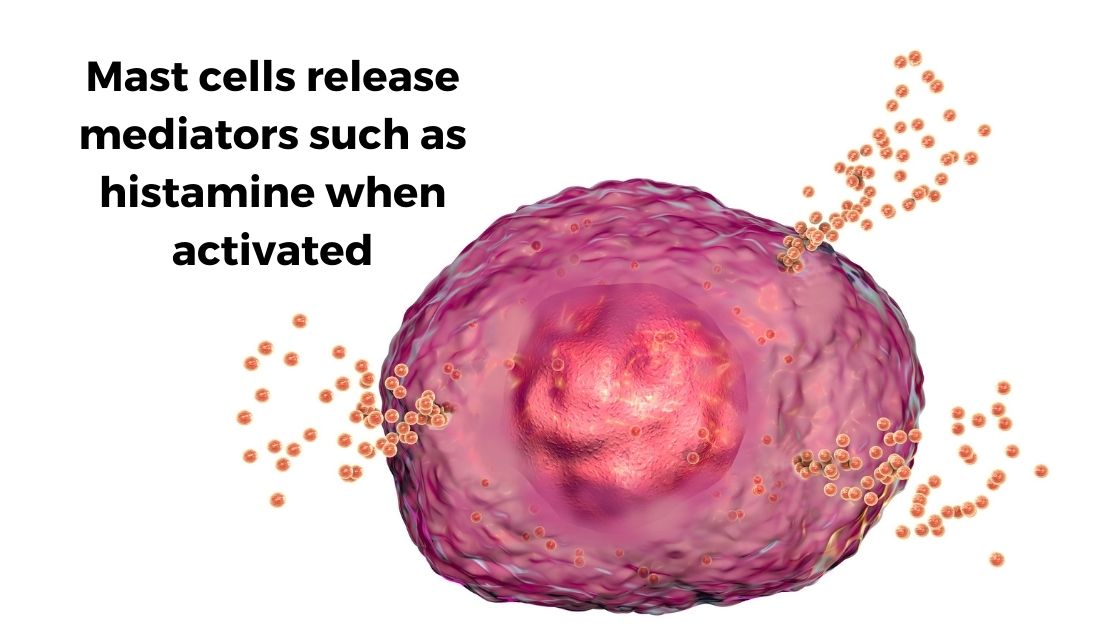
Mast cells are also important in wound healing and repair, and they are found in connective tissue. Mast cells are found abundantly in the skin, gastrointestinal tract, and respiratory tract. Additionally, mast cells can modulate heart rate by releasing histamine or tryptase.[ref]
Overlaps between mast cells and hypermobility Ehlers-Danlos syndrome:
- In the joints, mast cells are an integral part of osteoarthritis. Tryptase released from mast cells induces inflammation and causes cartilage breakdown.[ref]
- In skin scarring, mast cells may also play an important role. Many studies show a higher number of mast cells or mast cell activation in scars.[ref]
- In tendon injuries, mast cells play an important role in modulating inflammation, pain, and healing. Mast cells are found not only in tendon injuries but also near peripheral nerves and may be involved in pain communication.[ref]
- Higher tryptase levels were found in a study of 35 families with skin and connective tissue abnormalities.[ref]
Related article: Read more about mast cell activation syndrome and genetics.
Fibromyalgia or ME/CFS and Ehlers-Danlos Syndrome:
Some patients with Ehlers-Danlos syndrome are misdiagnosed as having either fibromyalgia or ME/CFS. In patients (without genetic mutations in the collagen genes) who are diagnosed with hEDS or hypermobility spectrum disorder, fibromyalgia or chronic fatigue may be coexisting conditions.[ref]
A study involving 63 fibromyalgia or ME/CFS diagnosed patients found 81% met the criteria for hypermobility syndrome and 18% met the criteria for hEDS.[ref]
Clinicians reported in the journal Clinical and Experimental Rheumatology that most of their hEDS patients also met the criteria for CFS.[ref]
Long Covid, ME/CFS, and hEDS:
About 30–57% of patients with long COVID, ME/CFS (chronic fatigue syndrome), fibromyalgia, or POTS also have symptoms of hEDS. Long Covid symptoms often include musculoskeletal pain, disautonomia, brain fog, and fatigue — which overlap with hEDS.[ref]
Another commonality between long Covid and hEDS is mast cell activation, which results in damage to the connective tissue. A case series detailing the overlap notes that the persistence of a viral infection can cause mast cell activation and hyperinflammation in the connective tissue. [ref]
Some researchers are now noting that ME/CFS patients with joint hypermobility often have more severe symptomatology and reduced quality of life.[ref]
Ehlers-Danlos Genotype Report
A couple of notes before you check your data:
- 23andMe and AncestryDNA raw data files are not guaranteed to be clinically accurate for medical diagnosis. False positives are possible, and anything you discover here should be replicated with a clinically accurate test.
- Mistakes are possible in the list below – it’s long, I’m human. Please contact me if you see anything listed incorrectly.
- This is only a portion of the known EDS mutations. If you or your doctor think that you have Ehlers-Danlos syndrome, you should seek a clinical test that covers all the mutations.
COL5A1 gene – Classical EDS rare mutations:
Access this content:
An active subscription is required to access this content.
Lifehacks: Natural Solutions for EDS that are Backed by Research
Real help from people who have been there:
Before I explain the research studies on possible solutions to help with Ehlers-Danlos symptoms, I want to share a good resource for anyone needing help with navigating EDS: Connective Tissue Coalition. From the site: “Connective Tissue Coalition (CTC) empowers individuals and families affected by Ehlers-Danlos Syndrome (EDS), Marfan Syndrome, and Loeys-Dietz Syndrome (LDS) through advocacy, research, and support.” Importantly, there is information on how to navigate the Social Security system in the US for getting disability.
Physical Interventions for EDS
Please talk with your EDS doctor before implementing any changes.
Access this content:
An active subscription is required to access this content.
Related Articles and Topics:
Histamine Intolerance: Genetic Report, Supplements, and Real Solutions
TNF-alpha: Inflammation, Chronic Diseases, and Genetic Susceptibility
Rheumatoid Arthritis: Genetics, Root Causes, and Treatment Research
References: