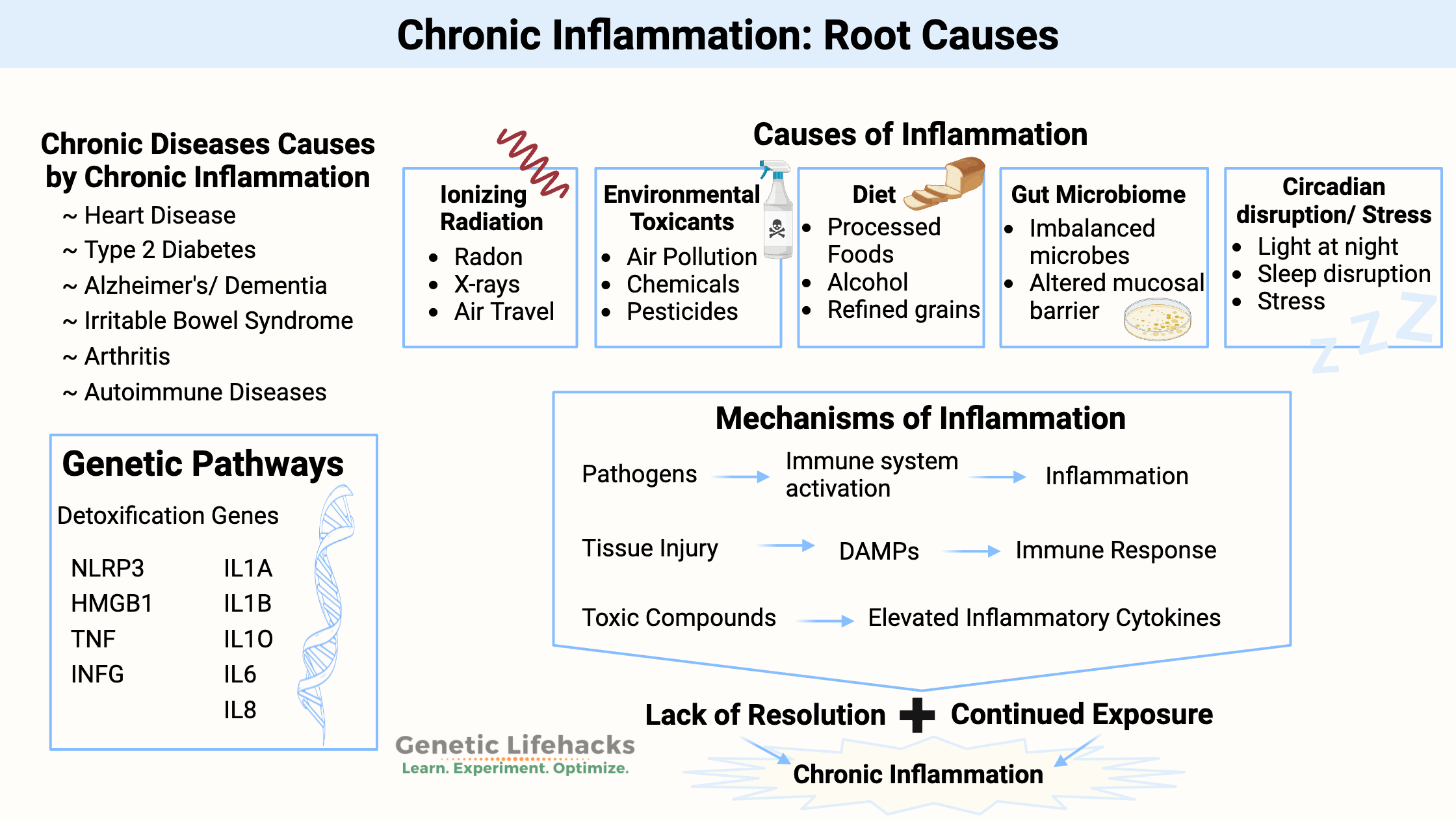
Key takeaways:
- Chronically elevated inflammatory cytokines are a primary root cause of chronic diseases such as Heart Disease, Alzheimer’s/Dementia, Autoimmune Diseases, Type 2 Diabetes, IBS, and Arthritis.
- For many people, specific inflammatory cytokines are likely to be higher due to genetic variants.
- Understanding these genetic pathways can lead you to the right solution for keeping inflammation under control.
What causes chronic inflammation?
Inflammation sometimes seems like a nebulous boogeyman talked about on health blogs without any specifics.
A popular health website states that inflammation is caused by: “A poor diet, stress, minor food allergies, a sedentary lifestyle, and more can contribute to chronic inflammation.”[ref]
But, my ‘poor diet’ is different than yours… and ‘minor food allergies’ just sounds made up to me!
Let’s lay this out logically and try to find some answers.
I’m going to focus on lifestyle, food, and environmental factors that cause elevated levels of inflammatory cytokines. These are the things you can optimize right now to impact chronic inflammatory diseases, along with the genetic variants that you need to understand and work with.
The purpose of understanding chronic inflammation is to prevent and/or reverse chronic diseases. Every time you see ‘chronic disease’, replace it with whatever condition you’re dealing with (e.g., joint pain, gum disease) or whatever you plan to avoid getting (e.g., diabetes or heart disease that runs in your family).
According to the NIH, the causes of chronic inflammation are:[ref]
- Environmental toxicants: chemicals we commonly encounter may alter the molecular pathways that underlie inflammation.
- Alterations to the gut due to diet: Diets high in refined grains, alcohol, and processed foods can alter the gut microbiota and lead to intestinal and immune system changes.
- Microbiome: An imbalance in the types of bacteria in the gut is linked to elevated inflammatory levels.
- Circadian disruption, Stress: Artificial light at night, sleep disruptions, and psychosocial stress influence the immune system.
- Early developmental conditions: Childhood obesity, psychological stress, exposure to microbes in infancy, and prenatal conditions are linked to inflammation later in life.
We are all unique in our kryptonite. Everyone has a different susceptibility level to chronic inflammation due to specific sources. Yes, I’m talking about genetics. Your genetic variants make you either more susceptible or more resilient to various environmental insults.
We’ll come back to your genetic variants in a minute. First, I’ll explain common reasons for inflammation to give you some background on what goes wrong in chronic inflammatory conditions.
What does your immune system have to do with inflammation?
Inflammation is one way that your immune system responds to several types of harm.
Throughout human history, the immune system has been protecting us from tons of different pathogens (bacteria, viruses, yeasts, and parasites). Prior to antibiotics, pathogens were the major cause of death. Our history as a species is molded by the pathogens we have lived through.
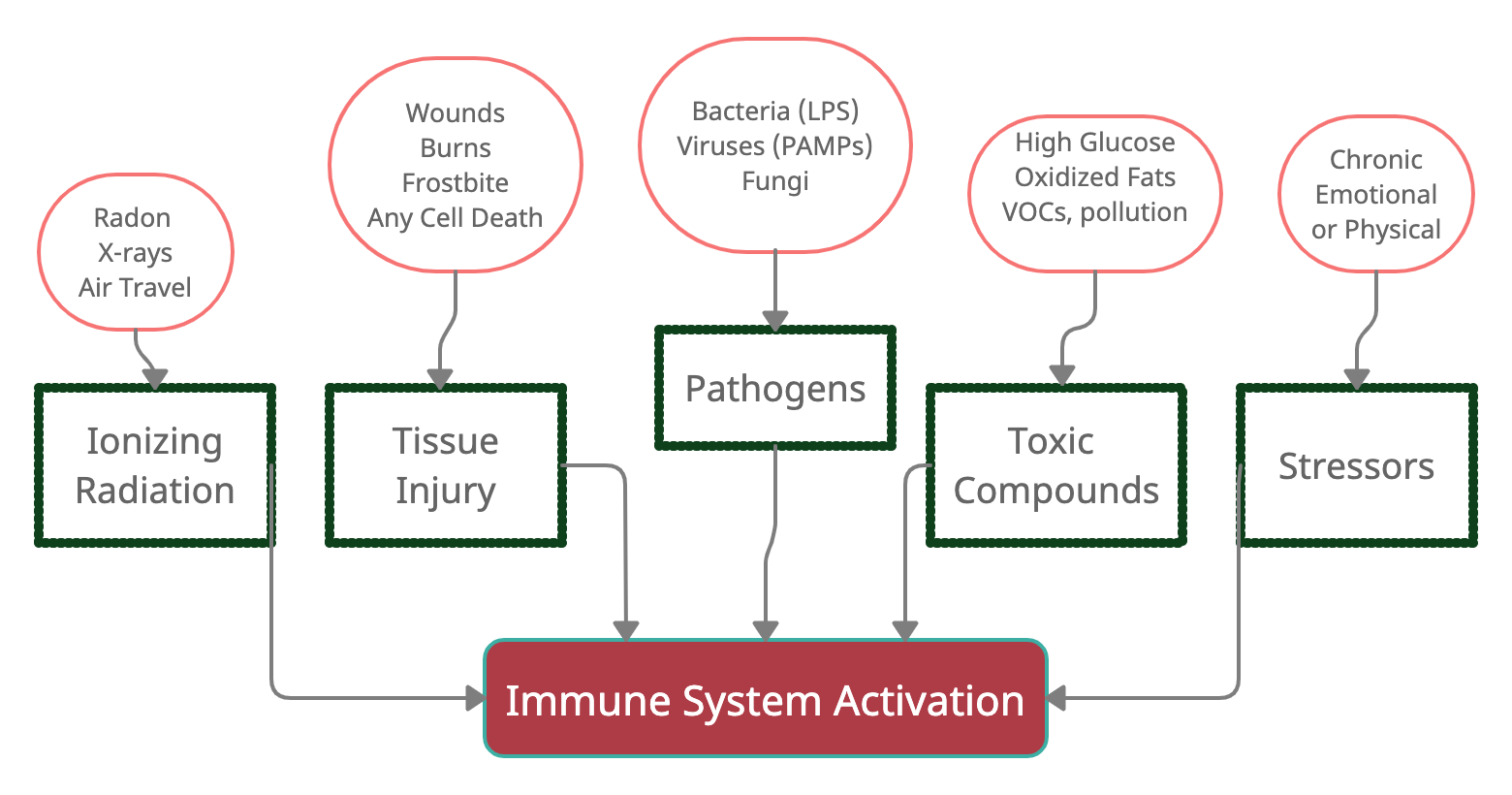
In general, the immune system responds to harmful stimuli, such as:[ref]
- pathogens (bacteria, viruses, fungi)
- tissue injury (wound, burn, injury, damage to cells)
- ionizing radiation (UV from the sun, x-rays)
- toxic compounds (man-made chemicals, alcohol, high blood glucose, heavy metals)
- psychological events (excitement) and physiological stress (marathon)
More on these in a minute…
Activating your immune system causes an inflammatory response, which has four components:
- inflammatory inducers
- sensors that detect the inflammatory inducers
- inflammatory mediators induced by the sensors
- target tissues affected by the inflammatory mediator.
This process looks different depending on what initiates the inflammatory response.
Example: When encountering a bacterial pathogen (inducer), the sensors are toll-like receptors on immune cells. The immune cells then express inflammatory mediators (TNF-alpha, IL-1, IL6, histamine, prostaglandins, etc.). The inflammatory mediators act on the target tissue, such as inducing vasodilation or recruiting white blood cells (neutrophils).[ref]
Different pathogens or toxins may induce different inflammatory mediators. For example, viral infections (inducers) prompt the release of interferon (mediator) from natural killer cells and T cells (sensors). Interferon then can interfere with viral replication or cause B cells (target) to produce antibodies.[ref]
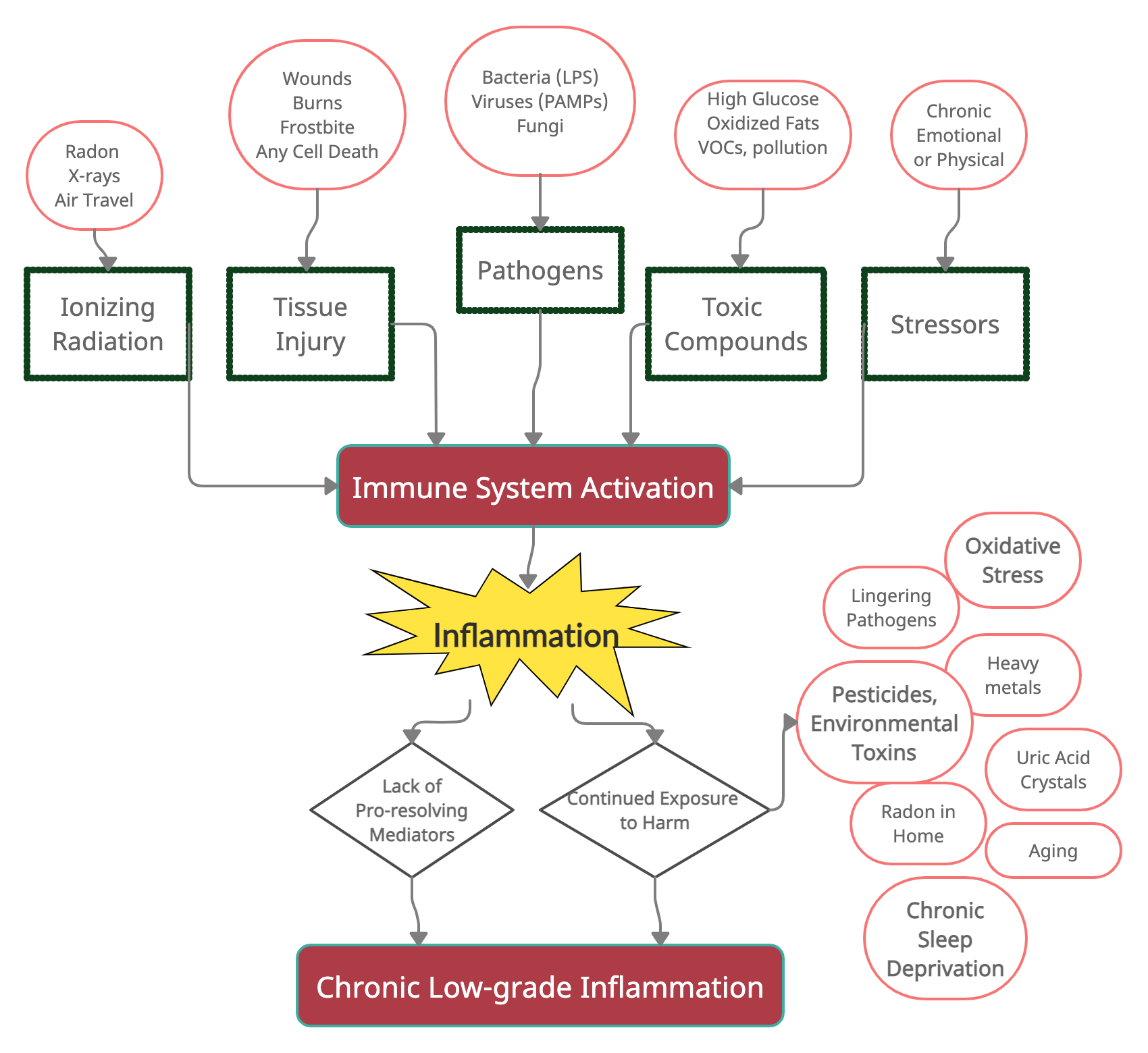
Here’s a graphical overview of what activates the immune response (I’ll add to this graphic as we go along).
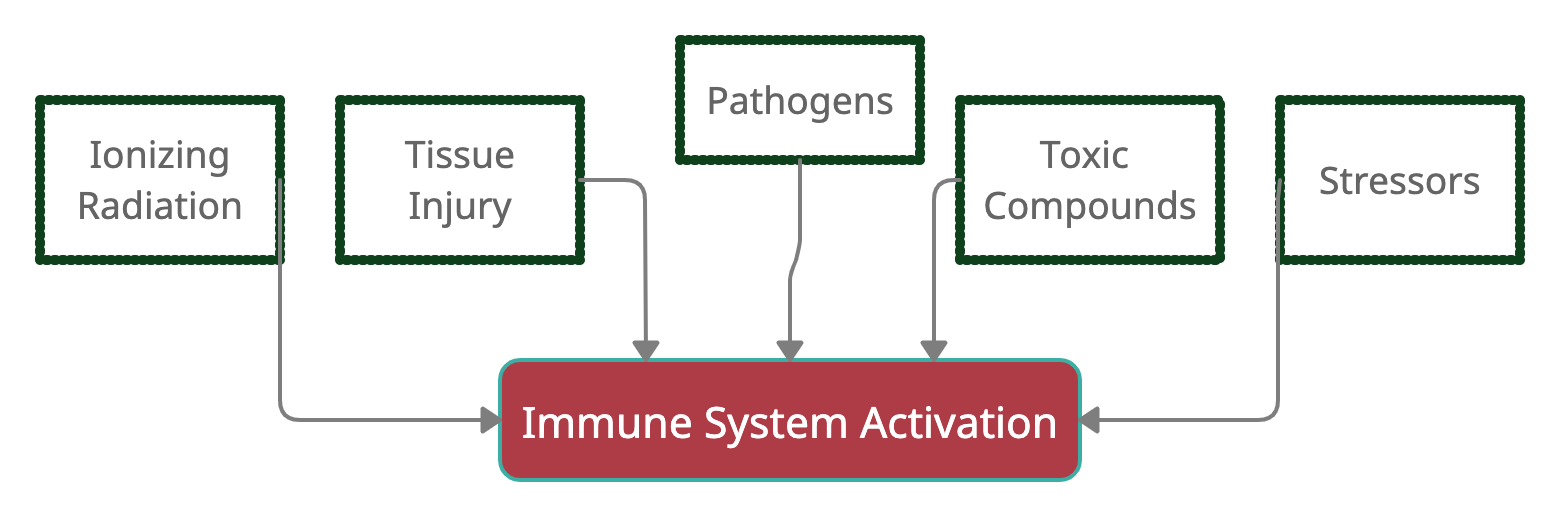
Let’s take a look at these inflammation activators in more detail:
1) Pathogens:
Our body has generalized ways of recognizing a ‘bad guy’ pathogen and initiating an immediate reaction. This initial response isn’t specific to a particular bacterial strain or virus — just a quick reaction to the detection of something likely ‘bad’.[ref]
- Bacteria with lipopolysaccharides (LPS) on the outer membrane trigger a specific response.
- Viruses trigger a different specific response based on recognition by PAMPs (pathogen-associated molecular patterns)
- The inflammatory response aims to protect the host from infection and then induce an adaptive immune response (e.g., make antibodies).
2) Tissue injury (burn, frostbite, injury, foreign body, trauma):
Inflammation is also caused by rupturing a cell membrane. This rupture could be due to a cut, bone break, crush wound, burn, or frostbite.
Anything that ruptures a cell causes the leakage of molecules in the cytosol of the cell – and some of the molecules trigger a ‘damage’ signal.
Essentially, the stuff inside a cell shouldn’t be found outside the cell.
Detection of DAMPs (damage-associated molecular patterns) triggers some of the same cascade of inflammatory events that detecting a pathogen – PAMPs – triggers. (Alarmin is another term used in research for DAMPs.)
The signals that activate DAMPs include:[ref][ref]
- molecules, such as ATP, released from dying cells
- breakdown products from the extracellular matrix
- products of proteolytic cascade released in vascular damage.
Tissue damage is detected by macrophages (a type of white blood cell) and by pain receptors. Toll-like receptors recognize some of the different molecules and then activate the immune response.
3) Damage from ionizing radiation:
X-rays, background atmospheric radiation, and radon gas are sources of ionizing radiation. Obviously, we are all exposed to some background radiation constantly, and the body is made for handling this.
Ionizing radiation releases energy in a way that is powerful enough to break chemical bonds. It can cause tissue damage, especially at higher levels.
At low levels, the problem with ionizing radiation is that it causes breaks in DNA strands. If the breaks aren’t repaired – or if they are repaired improperly – DNA sequence changes (mutations) can result, which could cause cancer.[ref]
Similar to DAMPs (damage-associated molecular patterns), when DNA is broken, a DNA damage response is activated. If the DNA damage is extensive, the cell should undergo apoptosis (cell death), which causes activation of the inflammatory system.[ref]
At low doses, ionizing radiation can upregulate inflammatory markers. It’s a fine balance, though. Really, really low doses of radiation prompt just enough response to have an overall anti-inflammatory effect.[ref][ref]
4) Toxic compounds:
Both endogenous (made in the body) and exogenous (from outside) compounds can be toxic or harmful and trigger an inflammatory response.
Examples include:
- high blood glucose levels
- oxidized fats, such as omega-6 fatty acids that are easily oxidized
- toxins (pesticides, plastics, flame retardants, VOCs, etc.)
- chemical irritants (fluoride, nickel, other trace elements)
High blood glucose:
Research shows that acutely raising blood glucose for several hours causes inflammatory cytokine (IL-6, TNF-alpha, and IL-18) levels to rise within two hours. The inflammatory cytokine levels returned to normal as blood glucose levels returned to baseline.[ref] While pre-diabetes may sound innocuous, the elevation in blood glucose causes a continual elevation in inflammatory markers.[ref]
Oxidized omega-6 fatty acids:
Over the past 70 years, one significant dietary change has been the increased consumption of omega-6 oils (corn, soybean, canola oil). It has been a huge shift away from what our ancestors ate. The omega-6 oils are easily oxidized, and the oxidized fatty acids trigger macrophages (white blood cells) which release inflammatory cytokines. While these omega-6 oils are labeled ‘heart healthy’ because they reduce LDL cholesterol, new research shows a different story. A British Medical Journal article concludes: “In summary, numerous lines of evidence show that the omega-6 polyunsaturated fat linoleic acid promotes oxidative stress, oxidised LDL, chronic low-grade inflammation and atherosclerosis, and is likely a major dietary culprit for causing CHD, especially when consumed in the form of industrial seed oils commonly referred to as ‘vegetable oils’.”[ref]
Chemical toxin exposure:
This topic is enormous, and many volumes could be written about it. Let me give you just one concrete example (with more in the following sections). Volatile organic compounds (VOCs) are released from many building materials, including paint, new furniture, perfumes, and petroleum products. Serum inflammatory cytokine levels (IL8, IL13, IL6, IL4, TNF-alpha) rise in conjunction with indoor VOC exposure.[ref]
Irritants:
Exposure to certain metals can trigger an inflammatory response in some people. For example, nickel is a common culprit in people with allergic dermatitis. Research shows that exposure to nickel increases TNF-α, IFN-γ, IL-4, and IL-13.[ref] Another example is titanium dental implants. Inflammation can increase due to interactions with toll-like receptors and titanian ions. Fluoride interacts with the titanium causing the release of titanium ions.[ref]
5) Psychological and physiological causes of inflammation:
Stress/Emotions:
Inflammatory markers elevate during stressful situations – and situations of “emotional attention”. It is one cause of depression.[ref] This seems to be a feedback loop. An increase in inflammatory cytokines is linked to an increase in emotional attention via the direct stimulation of cortisol and ACTH from cytokine signaling.[ref]
Excessive exercise:
Endurance exercise, such as marathon running, causes increases in IL1, IL6, and IL8 for hours after exercise.[ref]
Here’s where we are so far:
What goes wrong to cause chronic, low-grade inflammation?
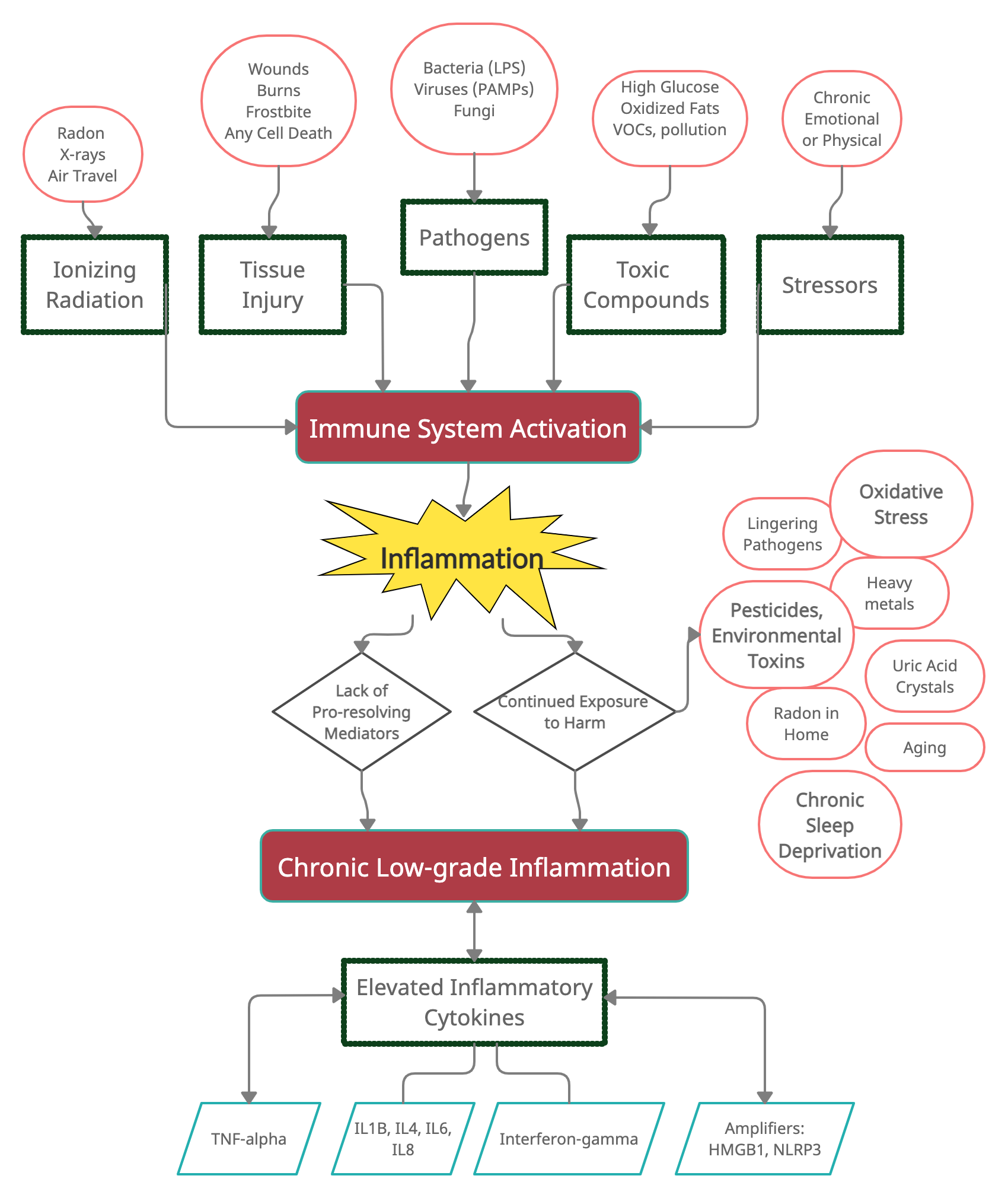
Theoretically, the immune system responds strongly to harmful stimuli (germs, wounds, toxic compounds, radiation), and then the system for the resolution of inflammation kicks in to bring everything back to normal.
I have another (long) article on the resolution of inflammation. In a nutshell, the resolution of inflammation is an active process that decreases inflammatory cytokine production alongside promoting healing processes (e.g., stem cells for repairing tissue).
The processes for resolving inflammation hinge on the formation of pro-resolving lipid mediators, which utilize DHA and EPA for biosynthesis.
At least half the picture in chronic inflammation is the lack of resolution of inflammation.
The other part of the picture here is continued exposure to harmful stimuli.
In my mind, it is an equation that looks like this:

One study puts it: “If the inflammatory trigger is not eliminated by the acute inflammatory response or persists for any other reason, the resolution phase may not be appropriately induced and a chronic inflammatory state may ensue. This state can be caused by chronic infections, unrepaired tissue damage, persistent allergens, undigestable foreign particles, or endogenous crystals, such as monosodium urate.”[ref]
As we talk about chronic inflammation, keep in mind that inflammatory cytokines can provoke cell death. While cell death occurs all the time in the body as cells reach the end of their replicative life, the type of cell death caused by inflammation is damaging. Cell death needs to be regulated — excessive cell death isn’t good.[ref][ref]
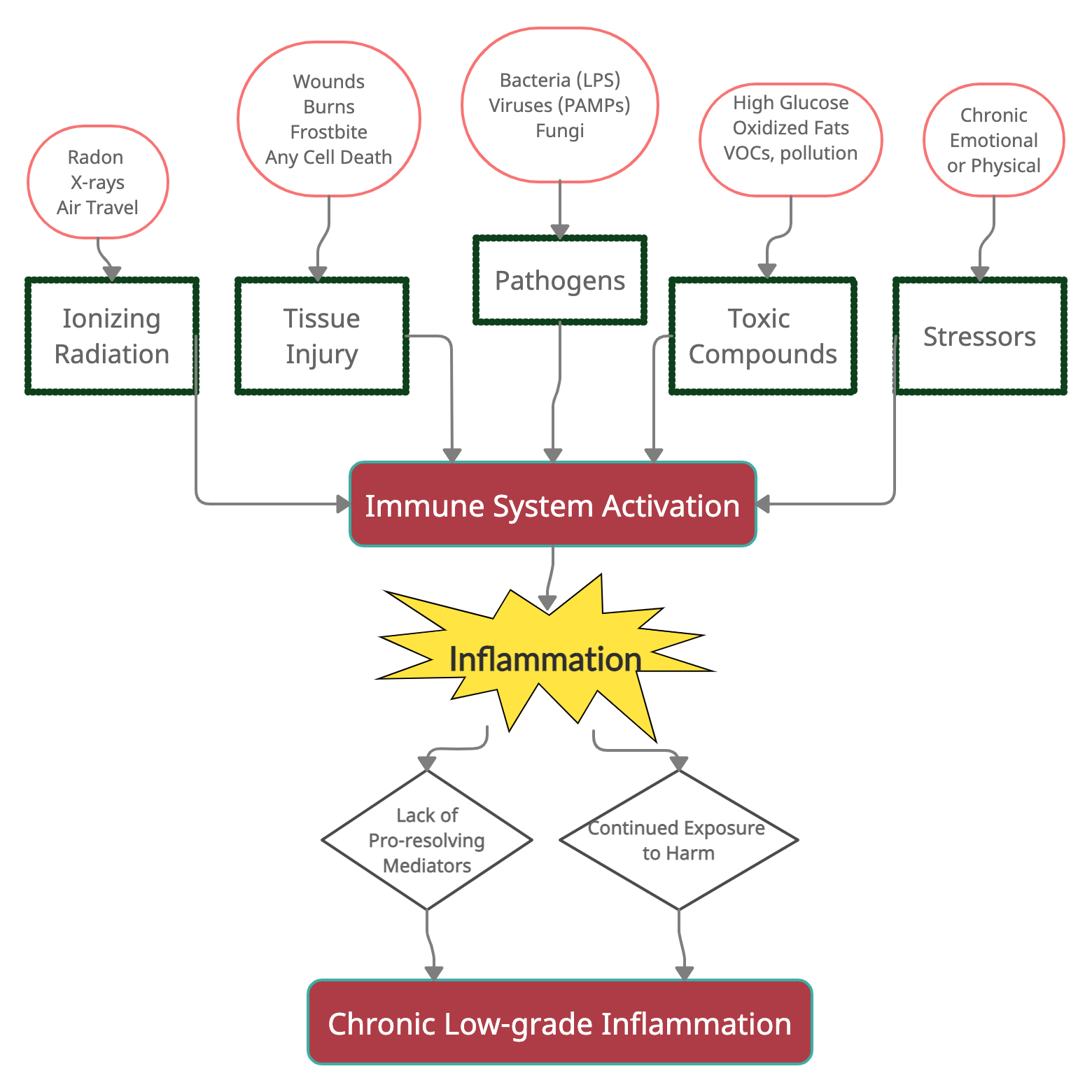
Let’s dive into the continued exposure to harmful stimuli part of the equation…
How can pathogens cause chronic inflammation?
Sometimes the immune system doesn’t clear out an infectious pathogen. The pathogen lingers at low levels, causing tissue-specific inflammation.
Here are several examples:
- P. gingivalis is a source of chronic inflammation in the gums (gingivitis). It also can travel in the bloodstream from your bleeding gums and is found in atherosclerotic plaque (linking it to heart disease).[ref]
- Epstein-Barr virus remains in the body for a lifetime. For most people, it is latent and does not cause problems. Some people, though, have a reactivation of the virus. It can be a cause of chronic fatigue[ref] and was recently linked to multiple sclerosis.[ref]
- Lyme disease is a tick-borne illness that can result in long-term, chronic inflammation for some people.
- Propionibacterium acnes is one skin bacteria that is linked to acne (a chronic inflammatory condition). But researchers have also found that Propionibacterium acnes may literally be at the heart of back pain (another chronic inflammatory condition). In a study on patients undergoing surgery for disc herniation, the researchers found that 44% of disc samples contained colonizing bacteria. P. acnes was the most common culprit by far. The bacteria formed a biofilm within the disc matrix and may be the source of inflammation in back pain for some people.[ref]
The gut microbiome and chronic inflammation:
Your intestinal barrier is amazing. It is a multilayer of defense to keep out pathogens, while simultaneously allowing for the absorption of water and nutrients.
Lining the epithelial cells of the intestines is a mucosal barrier. When the mucosal barrier is breached, and pathogens attack endothelial cells, there is an immediate and localized inflammatory response. Ideally, the immune system takes care of the pathogen, and the resolution of inflammation processes will return the cells to normal. But when too much damage or chronic barrier damage occurs, the inflammatory response becomes systemic. Too much immune response in intestinal endothelial cells can lead to further intestinal permeability, more pathogen and food protein infiltration, and chronic inflammation.[ref]
Another cause of chronic inflammation in the intestines is an overgrowth of ‘bad’ bacteria. One example of this is an infection with C. difficile. A C. diff infection causes direct injury to the cells lining the intestines. It causes the release of inflammatory cytokines, which, when overactive, cause further damage to intestinal cells.[ref]
Certain gut microbes also can stimulate inflammatory cytokine production. For example, Candida albicans and certain Bacteroides species are linked with higher levels of IL1B, IL6, and TNF-alpha.[ref]
Continued exposure to toxins:
I’m going to hit the highlights here to illustrate the point that different environmental toxins increase specific inflammatory cytokines in an individual manner.
Mercury:
Methylmercury exposure at low levels increases inflammatory cytokines.[ref] Methylmercury is formed by microbes in aquatic systems, which is why eating fish is a large source of methylmercury exposure. Industrial pollution can also be a source of methylmercury.
Methylmercury increases TNF-alpha expression. While methylmercury can accumulate in various tissues, the highest levels of TNF-alpha caused by methylmercury were found in the brain. It may be why mercury selectively damages neuronal tissue (via high TNF-alpha).[ref]
Lead:
Exposure to lead in drinking water increases inflammatory cytokines.[ref] Lead can be absorbed and stored in various organs. This storage can cause issues later in life, such as during kidney dialysis. People undergoing dialysis are at a significantly increased risk of carpal tunnel syndrome, an inflammatory condition. Dialysis can concentrate lead that is being excreted. It increases lead levels, and the increase in inflammation causes carpal tunnel syndrome.[ref]
Higher blood levels of lead, cadmium, and arsenic are directly associated with higher TNF-alpha levels.[ref]
Air pollution:
Breathing in urban air inevitably means taking in fine particulate matter. Urban air pollution causes the lungs to release inflammatory cytokines, including IL-6.[ref]
Pesticides:
Exposure to pesticide residue on fruits and vegetables is almost unavoidable in our modern world and a continual, small source of inflammation. For example, neonicotinoid pesticide residue is found on commonly consumed foods such as cherries, apples, pears, strawberries, cauliflower, cilantro, grapes, and leafy greens. Sometimes even in products labeled as organic.[ref] Exposure to neonicotinoids increases pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, IL-12, and IFN-γ.[ref]
Do you need to avoid all types of pesticides? Some people may be more affected by specific types of pesticides than others. Genetic variants are linked to increased negative effects of specific pesticides. I have free articles available explaining the specific variants associated with the following types of pesticides:
Arsenic:
While your first thought of arsenic may be as a poison used in mystery novels, arsenic is actually naturally occurring in groundwater in locations around the world. In high doses, arsenic kills everyone. At low-dose exposure through groundwater, arsenic detoxification uses up glutathione, which raises oxidative stress in cells. The increase in oxidative stress leads to chronic inflammation.
Related article: Arsenic detoxification genes
Uric acid:
Not all toxins come from outside the body. A byproduct of metabolizing purines (nucleic acids) is the creation of uric acid. Normally, uric acid is excreted in the urine.
Excessive uric acid can create urate crystals that deposit in different tissues (such as in the big toe in gout). However, uric acid is one of those danger-associated molecular patterns (DAMPs) that trigger an immune response.[ref]
What causes excess uric acid formation? A big reason for this excess in our modern diet is fructose. When high levels of fructose are consumed quickly (e.g., drinking a soda), the liver produces uric acid.[ref]
Related article: Uric acid genes
Plastics:
BPA and phthalates are two chemicals released from plastics and other products.
A study (in female mice) found that BPA at human-relevant doses increased inflammatory cytokines in the heart. When coupled with a viral infection, this increase increases the rate of myocarditis.[ref]
Related articles: BPA detoxification genes and Phthalate detoxification
Quinolone antibiotics as an environmental contaminant:
Antibiotics are used in huge amounts in conventional meat production. Environmental contamination (e.g., in the water supply or the soil as contamination from manure) means that we are exposed to low residual levels of quinolone antibiotics all the time. A recent study found that at environmental residual levels, quinolone antibiotics cause the increased secretion of pro-inflammatory cytokines.[ref]
Age-related chronic inflammation:
Chronic, low-level inflammation increases as we age.
When a cell reaches the end of its replicative ability, it goes into a state called senescence. Cellular senescence happens throughout life, and senescent cells are eventually removed via the immune system. In order to alert the immune system that they need to be removed, senescent cells secrete pro-inflammatory signals.
As we age, we end up with more senescent cells than the body can get rid of. Thus, we end up with cells that give off inflammatory cytokines, which then cause surrounding cells to become damaged.
Some researchers refer to this excess of inflammation due to senescent cell buildup in aging as “inflammaging”. Essentially, too many senescent cells cause chronic, low-grade inflammation in aging.[ref]
Related article: Senescence and telomeres
The second source of chronic inflammation in aging is due to glycosylated proteins, which are proteins that have been permanently altered. One source is advanced glycation end-products (AGEs), which build up in aging.[ref]
Related article: Advanced glycation end products
Radiation exposure:
You may think you don’t have to worry about radiation… as long as a nuclear bomb doesn’t go off or a nuclear plant doesn’t melt down.
But the second leading cause of lung cancer (after cigarettes) is radon exposure. Radon is a radioactive chemical found naturally in the environment in small amounts. It is gaseous and comes from the ground in varying amounts depending on geography. If you have radon in your area, radon test kits and meters are available to monitor the levels in your home.
Some people are better than others at mitigating the effects of radon. Certain genetic variants increase the risk of lung cancer from radon by 3-fold.[ref]
High levels of ROS: Oxidative Stress, Inflammation, Cell Death
Reactive oxygen species (ROS) are the byproducts of oxygen metabolism. All of your cells create a little ROS when creating energy in the mitochondria. And normal amounts of ROS are necessary and used for cell signaling.
Examples of ROS include hydrogen peroxide, superoxide, and hydroxyl radicals. All of these play a normal role in cell function when at low amounts.
The body’s natural antioxidant system prevents excesses… or at least it is supposed to. A decreased ability to form endogenous antioxidants (glutathione, superoxide dismutase, or catalase) can result in excess ROS. Genetics and nutrition both play a significant role in your cells’ ability to produce these essential antioxidants.
Excessive ROS production and/ or not enough antioxidants lead to oxidative stress. (I often think of oxidative stress as similar to iron rusting when left outside or an apple turning brown when exposed to the air — the same type of oxidation process.)
Important here: Oxidative stress leads directly to inflammatory cytokine creation in the cells. As one research paper puts it: “…oxidative stress can lead to chronic inflammation, which in turn could mediate most chronic diseases, including cancer, diabetes, cardiovascular, neurological and pulmonary diseases.”[ref]
Exposure to cigarette smoke, pollution, and heavy metals can cause oxidative stress in cells when the natural antioxidant system is overwhelmed. Excess ROS (oxidative stress) can cause DNA damage, which then triggers the activation of inflammatory pathways.
At first glance, this may seem like just another way that toxins cause chronic inflammation. But oxidative stress is also caused by radiation, high blood glucose, alcohol, and too much iron. Additionally, sources of chronic inflammation, such as obesity, atherosclerosis, and cancer, can also increase oxidative stress in cells. All of this triggers the activation of inflammatory pathways — and then certain immune system cells also create ROS as a way to create apoptosis. It is like a positive feedback loop with inflammation-causing ROS and then excessive ROS causing more inflammation.[ref]
Sleep deprivation, ROS in the intestines, and early death
Did you know that you can go longer without food than without sleep? All animals sleep, and sleep deprivation will kill any animal after a certain amount of time. But why?
Researchers did a bunch of experiments using model animals to determine which systems in the body were most affected in the body by lack of sleep. During sleep, cells repair. It was theorized that without sleep, cells in the brain would have increased oxidative stress, which would result in brain cell death.[ref]
But that isn’t what the researchers found. It turns out that with sleep loss, increased ROS in the gut causes oxidative stress and inflammation in the intestinal cells.
The good news here is that stopping the sleep loss eventually reversed the oxidative stress, and the previously sleep-deprived animals would live just as long as normal.
But wait, there’s more…
The other fascinating finding in the study was that feeding the animals antioxidants both reversed the buildup of ROS in the intestines. The animals given certain antioxidants (melatonin, lipoic acid, or NAC) survived a normal lifespan, even with severe sleep deprivation.[ref]
Here’s where we are now:
Naming Names: What do the specific inflammatory cytokines do?
I keep mentioning inflammatory cytokines, such as TNF-alpha and IL-6, without fully explaining them.
Cytokines are small proteins that act as signaling molecules. There are a bunch of different inflammatory cytokines, and they are released from different types of immune cells, like macrophages, mast cells, and T cells.
As signaling molecules, they attach to receptors on cell membranes and cause a cascade of events to occur within a cell. While each cytokine and receptor combination causes a unique set of events to occur, in general, cytokines cause cells to upregulate inflammatory genes.
When you read about cytokines, you may think that several of them do pretty much the same thing. There does seem to be a lot of redundancy in this system — which is good since you rely on cytokines for fighting off pathogens on an ongoing basis.
On the other hand, the excessive inflammatory response has a negative effect on tissue function and can cause overt tissue damage.
To sum up: cytokines are released by immune cells, attach to surface receptors on other cells, and cause the cell to produce an inflammatory response.
Summary of cytokines and their functions (CC source).[ref]
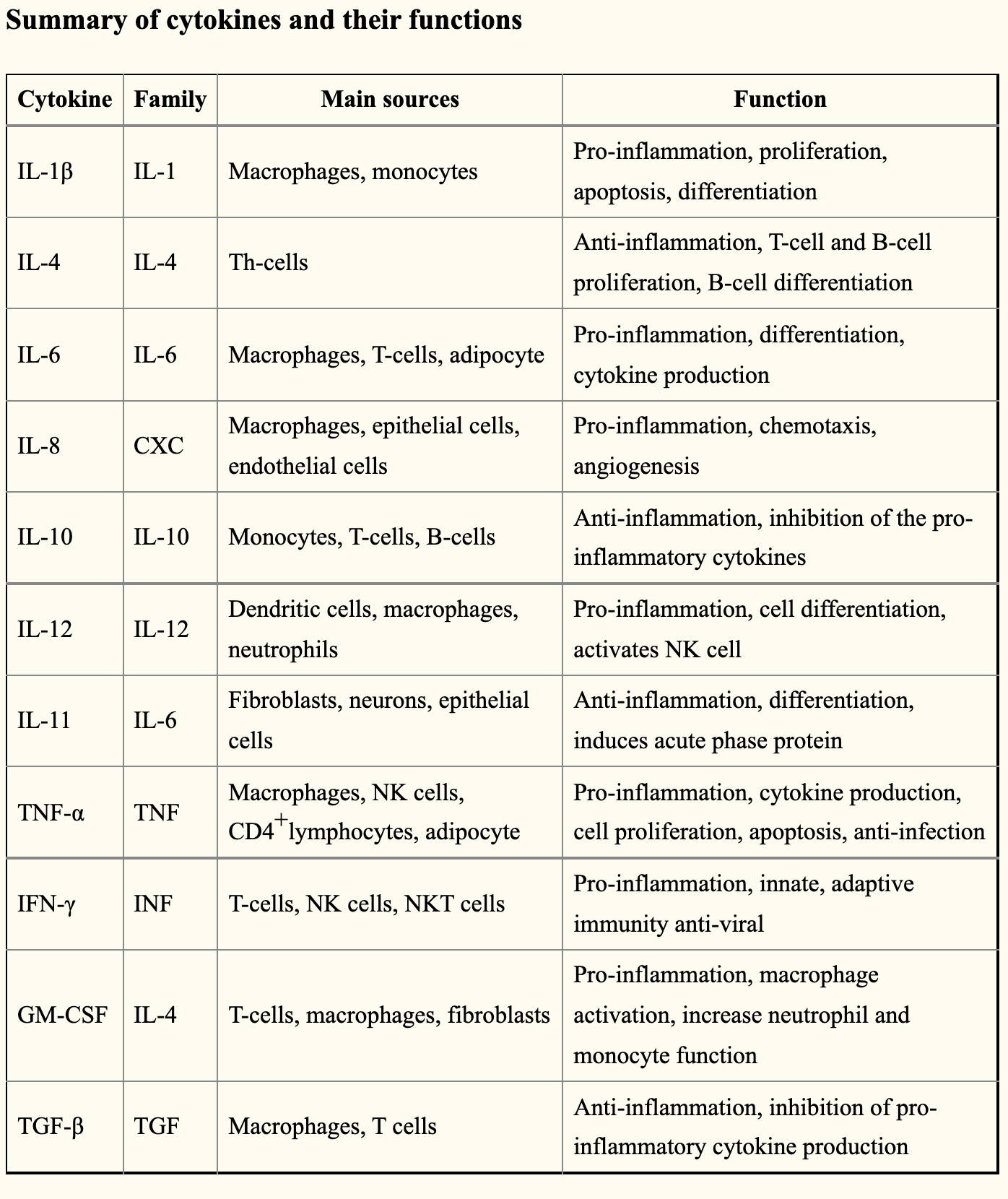
I’ll cover a number of these cytokines below in the genetic variants section.
Inflammatory proteins and enzymes:
Acute inflammatory proteins can be a marker of inflammation in the body. These inflammatory proteins include C-reactive protein (CRP), fibrinogen, haptoglobin, and serum amyloid A. During trauma, stress, or infection, these inflammatory proteins are elevated and try to restore the body or tissue to normal.[ref]
Related articles: C-Reactive Protein Genes, Fibrinogen
Amplifying the signal: HMGB1
HMGB1 is a multipurpose protein. Normally, it resides in the cell nucleus and helps to organize DNA for transcription. But when there is a pathogen or high levels of ROS, HMGB1 moves into the cytosol of the cell. It can act to activate a feedback loop that increases the production of several inflammatory cytokines.[ref]
Related article: HMGB1 Gene and Inflammatory Response
NLRP3 Inflammasome:
Another way to ramp up the inflammatory response system is through activation of NLRP3.
Related article: NLRP3 inflammasome
Exposure to environmental toxicants can result in organ damage or autoimmune diseases. “The exaggerated activation of the NLRP3 inflammasome and its end product IL-1β, is responsible for the pathogenesis caused by many ETs including pesticides, organic pollutants, heavy metals, and crystalline compounds.”[ref]
Naming names on what activates NLRP3: Paraquat (herbicide), rotenone (pesticide), chlorpyrifos (organophosphate pesticide), phthalates (plasticizers), cadmium, chromium, mercury, nickel (e.g., from jewelry and clothing buttons), silica, and asbestos.
High homocysteine levels activate the NLRP3 inflammasome. (I’ll come back to this in the genetics section with MTHFR and the lifehacks section with folate.)[ref]
Related article: Homocysteine – genetics and solutions
Genotype Report: Inflammation
Access this content:
An active subscription is required to access this content.
Lifehacks:
Here’s the stuff you’ve been waiting for — the scoop on specific ways of targeting chronic inflammation, based on your genes.
Diet:
Cut out processed junk foods:
In general, eating ultra-processed foods is linked to increased pro-inflammatory biomarkers (CRP, IL6, TNF-alpha).[ref] Ultraprocessed foods come in a box at the grocery store — cookies, candies, cereal, frozen pizza, chicken nuggets, corn dogs, etc.
So what is it about eating a lot of ultra-processed foods that cause inflammation? A 2021 research paper compiled data from hundreds of other studies. Surprisingly (to me), neither higher sugar nor lower fiber intake were consistently associated with inflammatory markers. Instead, it looks like the positive benefits of vitamins and antioxidants from fruits, vegetables, and quality meats (including fish) are decreasing inflammatory cytokines. The negatives of ‘ultra-processed foods’ in the Western diet may hinge on the high levels of omega-6 fats and the low intake of omega-3s.[ref] Omega-6 fats are easily oxidized, and the lack of omega-3 DHA and EPA means not enough pro-resolving mediators to turn off inflammatory processes.
Consume DHA and EPA:
Found in fish and in grass-fed meat, omega-3 fatty acids are the precursors for the pro-resolving mediators that turn off inflammation and restore tissue homeostasis. (Seriously, go read about specialized pro-resolving mediators, if you haven’t already.)
Fructose and Impaired Intestinal Barrier:
A diet that includes a lot of high-fructose corn syrup (animal study) causes leaky gut, which in turn increases inflammatory cytokines (TNF-alpha, IL-6). “the higher level intake of fructose impaired intestinal barrier function due to the decrease of the expression of tight junction proteins (ZO-1 and occludin).”[ref]
Emulsifiers and Impaired Intestinal Barrier:
Food additives such as emulsifiers, organic solvents, gluten, and nanoparticles used in processed food increase intestinal permeability. Essentially, they cause leaky gut, which increases inflammation through combating relocated gut bacteria.[ref]
Lifestyle factors:
Sleep Deprivation Kills You Via Increased ROS:
In case you skipped over it above, researchers have now figured out why sleep deprivation shortens lifespan (and total lack of sleep kills you quickly). The surprising result is that increased ROS (oxidative stress) in the gut is the culprit. Animal studies using a couple of different model animals all showed the same result – oxidative stress in the gut causes mortality in sleep deprivation.[ref]
Oxidative stress directly increases inflammatory cytokines. Importantly, early mortality due to sleep deprivation is reversed by antioxidants (melatonin, lipoic acid, or N-acetylcysteine).
Prioritize your sleep. Plan for enough time in bed; keep your room cool; sleep in the dark. Exposing your eyes to light in the blue spectrum (phones, TV, laptops) after dark decreases melatonin.
If you are regularly sleep deprived and can’t fix the source, consider whether melatonin supplements at night and either lipoic acid or N-acetylcysteine supplements in the daytime are appropriate for you.
23 Supplements that target specific inflammatory genetic pathways:
I’m going to list natural supplements that have been shown in research studies to impact specific inflammatory cytokines. There is some overlap with some supplements impacting multiple cytokines. Talk with your doctor if you have any questions about supplements and their interactions with prescription medications.
Access this content:
An active subscription is required to access this content.
Related Article:
Is Inflammation Causing Your Depression and Anxiety? The Science Behind the Link
Specialized Pro-resolving Mediators (SPMs): The Resolution of Inflammation
References:
Anderton, Holly, et al. “Cell Death in Chronic Inflammation: Breaking the Cycle to Treat Rheumatic Disease.” Nature Reviews Rheumatology, vol. 16, no. 9, Sept. 2020, pp. 496–513. www.nature.com, https://doi.org/10.1038/s41584-020-0455-8.
APA PsycNet. https://doi.apa.org/doiLanding?doi=10.1037%2Fa0035302. Accessed 4 Apr. 2022.
Audi, Christelle, et al. “Serum Cytokine Levels Related to Exposure to Volatile Organic Compounds and PM2.5 in Dwellings and Workplaces in French Farmers – a Mechanism to Explain Nonsmoking COPD.” International Journal of Chronic Obstructive Pulmonary Disease, vol. 12, May 2017, pp. 1363–74. PubMed Central, https://doi.org/10.2147/COPD.S117866.
Baselet, Bjorn, et al. “Pathological Effects of Ionizing Radiation: Endothelial Activation and Dysfunction.” Cellular and Molecular Life Sciences, vol. 76, no. 4, 2019, pp. 699–728. PubMed Central, https://doi.org/10.1007/s00018-018-2956-z.
Belli, Mauro, and Maria Antonella Tabocchini. “Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection.” International Journal of Molecular Sciences, vol. 21, no. 17, Aug. 2020, p. 5993. PubMed Central, https://doi.org/10.3390/ijms21175993.
Bruno, Katelyn Ann, et al. “BPA Alters Estrogen Receptor Expression in the Heart After Viral Infection Activating Cardiac Mast Cells and T Cells Leading to Perimyocarditis and Fibrosis.” Frontiers in Endocrinology, vol. 10, Sept. 2019, p. 598. PubMed Central, https://doi.org/10.3389/fendo.2019.00598.
Capoor, Manu N., et al. “Propionibacterium Acnes Biofilm Is Present in Intervertebral Discs of Patients Undergoing Microdiscectomy.” PLOS ONE, vol. 12, no. 4, Apr. 2017, p. e0174518. PLoS Journals, https://doi.org/10.1371/journal.pone.0174518.
Chen, Linlin, et al. “Inflammatory Responses and Inflammation-Associated Diseases in Organs.” Oncotarget, vol. 9, no. 6, Dec. 2017, pp. 7204–18. PubMed Central, https://doi.org/10.18632/oncotarget.23208.
Craddock, Hillary A., et al. “Trends in Neonicotinoid Pesticide Residues in Food and Water in the United States, 1999–2015.” Environmental Health, vol. 18, Jan. 2019, p. 7. PubMed Central, https://doi.org/10.1186/s12940-018-0441-7.
Ding, Cheng, et al. “TNF-α Gene Promoter Polymorphisms Contribute to Periodontitis Susceptibility: Evidence from 46 Studies.” Journal of Clinical Periodontology, vol. 41, no. 8, Aug. 2014, pp. 748–59. PubMed, https://doi.org/10.1111/jcpe.12279.
DiNicolantonio, James J., and James H. O’Keefe. “Omega-6 Vegetable Oils as a Driver of Coronary Heart Disease: The Oxidized Linoleic Acid Hypothesis.” Open Heart, vol. 5, no. 2, Oct. 2018, p. e000898. openheart.bmj.com, https://doi.org/10.1136/openhrt-2018-000898.
Durães, Cecília, et al. “Polymorphisms in the TNFA and IL6 Genes Represent Risk Factors for Autoimmune Thyroid Disease.” PloS One, vol. 9, no. 8, 2014, p. e105492. PubMed, https://doi.org/10.1371/journal.pone.0105492.
Duzguner, Vesile, and Suat Erdogan. “Chronic Exposure to Imidacloprid Induces Inflammation and Oxidative Stress in the Liver & Central Nervous System of Rats.” Pesticide Biochemistry and Physiology, vol. 104, no. 1, Sept. 2012, pp. 58–64. ScienceDirect, https://doi.org/10.1016/j.pestbp.2012.06.011.
Esposito, Katherine, et al. “Inflammatory Cytokine Concentrations Are Acutely Increased by Hyperglycemia in Humans: Role of Oxidative Stress.” Circulation, vol. 106, no. 16, Oct. 2002, pp. 2067–72. PubMed, https://doi.org/10.1161/01.cir.0000034509.14906.ae.
Feng, Bo, et al. “Association of Tumor Necrosis Factor α -308G/A and Interleukin-6 -174G/C Gene Polymorphism with Pneumonia-Induced Sepsis.” Journal of Critical Care, vol. 30, no. 5, Oct. 2015, pp. 920–23. PubMed, https://doi.org/10.1016/j.jcrc.2015.04.123.
Higo, Tomoaki, et al. “DNA Single-Strand Break-Induced DNA Damage Response Causes Heart Failure.” Nature Communications, vol. 8, Apr. 2017, p. 15104. PubMed Central, https://doi.org/10.1038/ncomms15104.
Houen, Gunnar, et al. “Epstein-Barr Virus and Multiple Sclerosis.” Frontiers in Immunology, vol. 11, Dec. 2020, p. 587078. PubMed Central, https://doi.org/10.3389/fimmu.2020.587078.
Huang, Wen-Hung, et al. “Blood Lead Level: An Overlooked Risk of Carpal Tunnel Syndrome in Hemodialysis Patients.” Renal Failure, vol. 41, no. 1, Nov. 2019, pp. 786–93. PubMed, https://doi.org/10.1080/0886022X.2019.1657894.
Huang, Yung-Cheng, et al. “Toxic Metals Increase Serum Tumor Necrosis Factor-α Levels, Modified by Essential Elements and Different Types of Tumor Necrosis Factor-α Promoter Single-Nucleotide Polymorphisms.” Epidemiology (Cambridge, Mass.), vol. 28 Suppl 1, Oct. 2017, pp. S113–20. PubMed, https://doi.org/10.1097/EDE.0000000000000738.
Hussain, Mehak, et al. “P. Gingivalis in Periodontal Disease and Atherosclerosis – Scenes of Action for Antimicrobial Peptides and Complement.” Frontiers in Immunology, vol. 6, Feb. 2015, p. 45. PubMed Central, https://doi.org/10.3389/fimmu.2015.00045.
Hussain, Tarique, et al. “Oxidative Stress and Inflammation: What Polyphenols Can Do for Us?” Oxidative Medicine and Cellular Longevity, vol. 2016, 2016, p. 7432797. PubMed Central, https://doi.org/10.1155/2016/7432797.
Iwai-Shimada, Miyuki, et al. “Methylmercury Induces the Expression of TNF-α Selectively in the Brain of Mice.” Scientific Reports, vol. 6, Dec. 2016, p. 38294. PubMed, https://doi.org/10.1038/srep38294.
Jim, Heather S. L., et al. “Genetic Predictors of Fatigue in Prostate Cancer Patients Treated with Androgen Deprivation Therapy: Preliminary Findings.” Brain, Behavior, and Immunity, vol. 26, no. 7, Oct. 2012, pp. 1030–36. PubMed, https://doi.org/10.1016/j.bbi.2012.03.001.
Khan, Saif, et al. “TNF-α -308 G > A (Rs1800629) Polymorphism Is Associated with Celiac Disease: A Meta-Analysis of 11 Case-Control Studies.” Scientific Reports, vol. 6, Sept. 2016, p. 32677. PubMed, https://doi.org/10.1038/srep32677.
Lang, Lang, et al. “Macrophage Polarization Induced by Quinolone Antibiotics at Environmental Residue Level.” International Immunopharmacology, vol. 106, Feb. 2022, p. 108596. PubMed, https://doi.org/10.1016/j.intimp.2022.108596.
Lorenzo-González, María, et al. “Residential Radon, Genetic Polymorphisms in DNA Damage and Repair-Related.” Lung Cancer (Amsterdam, Netherlands), vol. 135, Sept. 2019, pp. 10–15. PubMed, https://doi.org/10.1016/j.lungcan.2019.07.003.
MacIntyre, Elaina A., et al. “GSTP1 and TNF Gene Variants and Associations between Air Pollution and Incident Childhood Asthma: The Traffic, Asthma and Genetics (TAG) Study.” Environmental Health Perspectives, vol. 122, no. 4, Apr. 2014, pp. 418–24. PubMed, https://doi.org/10.1289/ehp.1307459.
Majumder, Poulami, et al. “Association of Tumor Necrosis Factor-α (TNF-α) Gene Promoter Polymorphisms with Aggressive and Chronic Periodontitis in the Eastern Indian Population.” Bioscience Reports, vol. 38, no. 4, Aug. 2018, p. BSR20171212. PubMed, https://doi.org/10.1042/BSR20171212.
Maydych, Viktoriya. “The Interplay Between Stress, Inflammation, and Emotional Attention: Relevance for Depression.” Frontiers in Neuroscience, vol. 13, 2019. Frontiers, https://www.frontiersin.org/article/10.3389/fnins.2019.00384.
Medzhitov, Ruslan. “Inflammation 2010: New Adventures of an Old Flame.” Cell, vol. 140, no. 6, Mar. 2010, pp. 771–76. www.cell.com, https://doi.org/10.1016/j.cell.2010.03.006.
Muniroh, Muflihatul. “Methylmercury-Induced pro-Inflammatory Cytokines Activation and Its Preventive Strategy Using Anti-Inflammation N-Acetyl-l-Cysteine: A Mini-Review.” Reviews on Environmental Health, vol. 35, no. 3, Sept. 2020, pp. 233–38. PubMed, https://doi.org/10.1515/reveh-2020-0026.
Netea, Mihai G., et al. “A Guiding Map for Inflammation.” Nature Immunology, vol. 18, no. 8, July 2017, p. 826. www.ncbi.nlm.nih.gov, https://doi.org/10.1038/ni.3790.
Nieman, David C., et al. “Cytokine Changes after a Marathon Race.” Journal of Applied Physiology, vol. 91, no. 1, July 2001, pp. 109–14. journals.physiology.org (Atypon), https://doi.org/10.1152/jappl.2001.91.1.109.
Pahwa, Roma, et al. “Chronic Inflammation.” StatPearls, StatPearls Publishing, 2022. PubMed, http://www.ncbi.nlm.nih.gov/books/NBK493173/.
Radbin, Rayhaneh, et al. “The Influence of Drinking-Water Pollution with Heavy Metal on the Expression of IL-4 and IFN-γ in Mice by Real-Time Polymerase Chain Reaction.” Cytotechnology, vol. 66, no. 5, Oct. 2014, pp. 769–77. PubMed Central, https://doi.org/10.1007/s10616-013-9626-7.
Reséndiz-Hernández, Juan Manuel, et al. “Identification of Genetic Variants in the TNF Promoter Associated with COPD Secondary to Tobacco Smoking and Its Severity.” International Journal of Chronic Obstructive Pulmonary Disease, vol. 10, 2015, pp. 1241–51. PubMed, https://doi.org/10.2147/COPD.S83298.
Reuter, Simone, et al. “Oxidative Stress, Inflammation, and Cancer: How Are They Linked?” Free Radical Biology & Medicine, vol. 49, no. 11, Dec. 2010, pp. 1603–16. PubMed Central, https://doi.org/10.1016/j.freeradbiomed.2010.09.006.
Sanada, Fumihiro, et al. “Source of Chronic Inflammation in Aging.” Frontiers in Cardiovascular Medicine, vol. 5, Feb. 2018, p. 12. PubMed Central, https://doi.org/10.3389/fcvm.2018.00012.
Schirmer, Melanie, et al. “Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity.” Cell, vol. 167, no. 4, Nov. 2016, pp. 1125-1136.e8. PubMed Central, https://doi.org/10.1016/j.cell.2016.10.020.
Shi, Yan. “Caught Red-Handed: Uric Acid Is an Agent of Inflammation.” The Journal of Clinical Investigation, vol. 120, no. 6, June 2010, pp. 1809–11. PubMed Central, https://doi.org/10.1172/JCI43132.
Silvestre, Marilene Chaves, and Vitor Manoel Silva Dos Reis. “Evaluation of the Profile of Inflammatory Cytokines, through Immunohistochemistry, in the Skin of Patients with Allergic Contact Dermatitis to Nickel in the Acute and Chronic Phases.” Anais Brasileiros De Dermatologia, vol. 93, no. 6, Dec. 2018, pp. 829–35. PubMed, https://doi.org/10.1590/abd1806-4841.20187126.
Soberanes, Saul, et al. “Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis.” Cell Metabolism, vol. 29, no. 2, Feb. 2019, pp. 335-347.e5. PubMed, https://doi.org/10.1016/j.cmet.2018.09.019.
Stappers, M. H. T., et al. “Polymorphisms in Cytokine Genes IL6, TNF, IL10, IL17A and IFNG Influence Susceptibility to Complicated Skin and Skin Structure Infections.” European Journal of Clinical Microbiology & Infectious Diseases: Official Publication of the European Society of Clinical Microbiology, vol. 33, no. 12, Dec. 2014, pp. 2267–74. PubMed, https://doi.org/10.1007/s10096-014-2201-0.
Tavares, M., et al. “Tumour Necrosis Factor-Alpha (-308G/A) Promoter Polymorphism Is Associated with Ulcerative Colitis in Brazilian Patients.” International Journal of Immunogenetics, vol. 43, no. 6, Dec. 2016, pp. 376–82. PubMed, https://doi.org/10.1111/iji.12289.
Thoo, Lester, et al. “Keep Calm: The Intestinal Barrier at the Interface of Peace and War.” Cell Death & Disease, vol. 10, no. 11, Nov. 2019, p. 849. PubMed Central, https://doi.org/10.1038/s41419-019-2086-z.
Vaccaro, Alexandra, et al. “Sleep Loss Can Cause Death through Accumulation of Reactive Oxygen Species in the Gut.” Cell, vol. 181, no. 6, June 2020, pp. 1307-1328.e15. www.cell.com, https://doi.org/10.1016/j.cell.2020.04.049.
Wachi, Takanori, et al. “Release of Titanium Ions from an Implant Surface and Their Effect on Cytokine Production Related to Alveolar Bone Resorption.” Toxicology, vol. 327, Jan. 2015, pp. 1–9. PubMed, https://doi.org/10.1016/j.tox.2014.10.016.
Wang, Zhen, et al. “Analysis of Inflammatory Mediators in Prediabetes and Newly Diagnosed Type 2 Diabetes Patients.” Journal of Diabetes Research, vol. 2016, July 2016, p. e7965317. www.hindawi.com, https://doi.org/10.1155/2016/7965317.
Wu, Jun-Cang, et al. “Gene Polymorphisms and Circulating Levels of the TNF-Alpha Are Associated with Ischemic Stroke: A Meta-Analysis Based on 19,873 Individuals.” International Immunopharmacology, vol. 75, Oct. 2019, p. 105827. PubMed, https://doi.org/10.1016/j.intimp.2019.105827.
Xu, Wen, and Yi Huang. “Regulation of Inflammatory Cell Death by Phosphorylation.” Frontiers in Immunology, vol. 13, 2022. Frontiers, https://www.frontiersin.org/article/10.3389/fimmu.2022.851169.
Yang, De, et al. “ALARMINS AND IMMUNITY.” Immunological Reviews, vol. 280, no. 1, Nov. 2017, pp. 41–56. PubMed Central, https://doi.org/10.1111/imr.12577.
Yu, Hua, et al. “Cytokines Are Markers of the Clostridium Difficile-Induced Inflammatory Response and Predict Disease Severity.” Clinical and Vaccine Immunology : CVI, vol. 24, no. 8, Aug. 2017, pp. e00037-17. PubMed Central, https://doi.org/10.1128/CVI.00037-17.
Zhang, Congwang, et al. “Recent Advances in Fructose Intake and Risk of Hyperuricemia.” Biomedicine & Pharmacotherapy, vol. 131, Nov. 2020, p. 110795. ScienceDirect, https://doi.org/10.1016/j.biopha.2020.110795.